ICU · Physiology / endocrine
Endocrine Physiology
Also known as Endocrine physiology · Hypothalamic-pituitary axis · HPA axis · Feedback loops · Thyroid physiology · Adrenal physiology · Glucose homeostasis · Calcium homeostasis · Sick euthyroid syndrome · Critical illness-related corticosteroid insufficiency · Vasopressin physiology · RAAS · Counter-regulatory hormones
Endocrine physiology: the hypothalamic-pituitary axes (the HPA — the CRH/ACTH/cortisol; the HPT — the TRH/TSH/T3/T4; the HPG — the GnRH/LH/FSH; the GH; the prolactin). The feedback loops (the negative). The adrenal (the cortisol, the aldosterone, the catecholamines). The glucose homeostasis (the insulin, the glucagon). The calcium homeostasis (the PTH, the vitamin D, the calcitonin). The clinical correlations (the thyroid storm, the adrenal crisis, the DKA, the SIADH).
On this page & tools
Your progress
Saved locally on this device.
8 MCQs with explanations
Target exams
Overview & definition
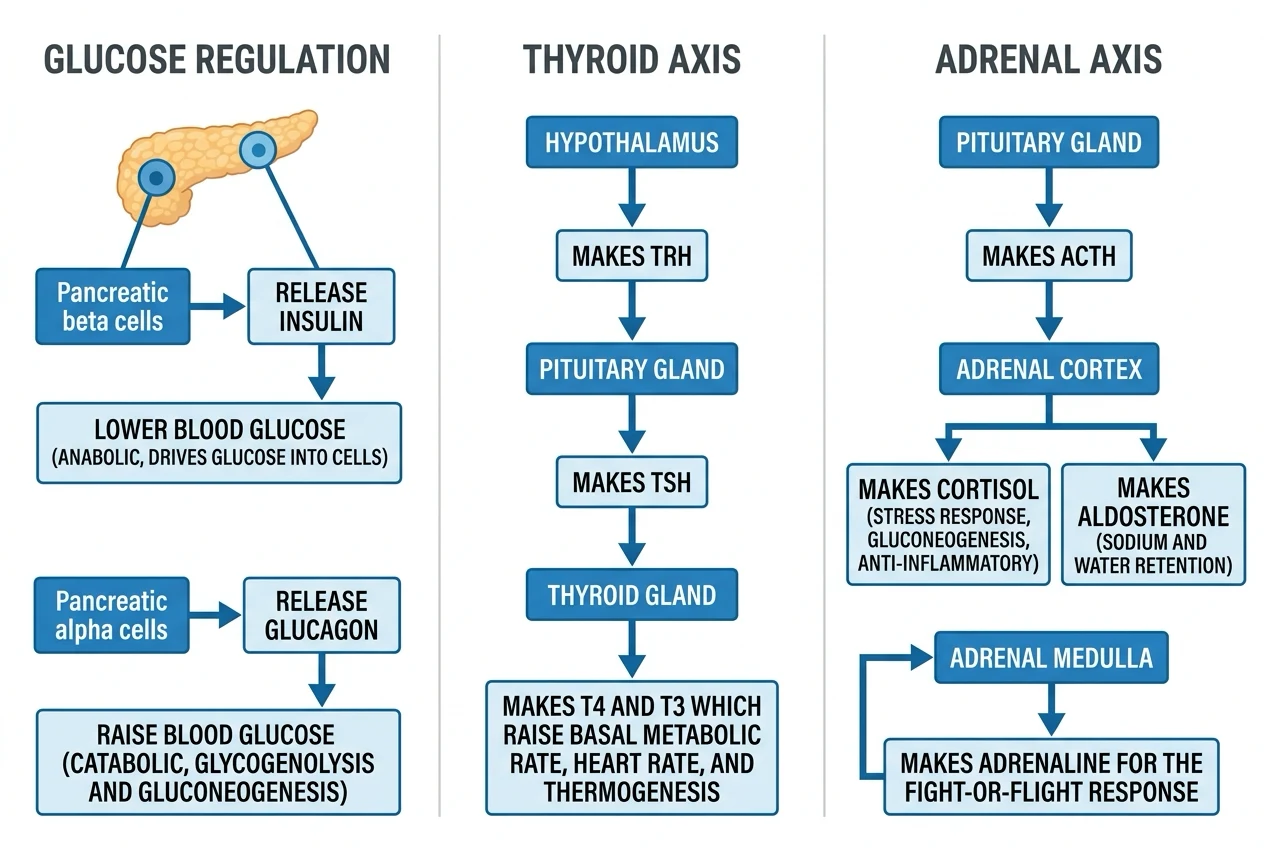
The endocrine physiology — the hypothalamic-pituitary axes (the master regulator). The feedback loops (the negative — the end-product inhibits the upstream). The adrenal, the thyroid, the pancreas (the glucose), the parathyroid (the calcium). The clinical correlations (the thyroid storm, the adrenal crisis, the DKA, the SIADH).[1]

Endocrine physiology matters in the ICU because critical illness is, in part, an endocrine event. Sepsis, trauma, burns and surgery are read by the hypothalamus as existential threats and re-programme every endocrine axis: cortisol and catecholamines rise to defend blood pressure, glucagon and counter-regulatory hormones drive stress hyperglycaemia, antidiuretic hormone retains water even at the cost of hyponatraemia, and thyroid hormone activation is deliberately switched off to conserve energy. The intensivist who understands these axes can distinguish an ADAPTIVE response (sick euthyroidism, stress hyperglycaemia) from a DEFICIENT one (critical illness-related corticosteroid insufficiency, relative vasopressin deficiency) — and treat only the latter.[5][9]
The axes


- The HPA (the hypothalamic-pituitary-adrenal): CRH → ACTH → cortisol (the negative the feedback). The cortisol (the stress the response; the - the - the).[1]
- The HPT (the hypothalamic-pituitary-thyroid): TRH → TSH → T3/T4 (the negative the feedback). The T4 (the - the - the); the T3 (the active; the - the - the).[1]
- The HPG (the hypothalamic-pituitary-gonadal): GnRH → LH/FSH → the testosterone/oestrogen.[1]
- The GH (the growth hormone) — the GHRH → GH → the IGF-1.[1]
- The prolactin — the - the - the (the dopamine the inhibits).[1]
Hypothalamic-pituitary axis — the master regulator
The hypothalamus is the central coordinator of the endocrine system. Neurosecretory cells in the paraventricular nucleus (and other hypothalamic nuclei) synthesise releasing and inhibiting hormones that travel via the hypophyseal portal venous system — a capillary bed at the median eminence that drains into a second capillary bed in the anterior pituitary. This portal arrangement delivers a high local concentration of hypothalamic hormones to the anterior pituitary while using very little total hormone — a feature that makes the axis both exquisitely sensitive and difficult to study with peripheral blood tests.[1]
The pituitary has two embryologically and functionally distinct parts. The anterior pituitary (adenohypophysis) arises from Rathke's pouch (oral ectoderm) and is a true endocrine gland whose cells synthesise and store hormones under hypothalamic control. The posterior pituitary (neurohypophysis) is a down-growth of neural tissue; it does not synthesise hormones but stores and releases oxytocin and vasopressin (ADH) made in hypothalamic neurons and transported down axons to terminal swellings (Herring bodies).[1]
The five hypothalamic-pituitary axes
| Axis | Hypothalamic hormone | Pituitary hormone(s) | End-organ | Feedback signal |
|---|---|---|---|---|
| HPA | CRH (+ ADH synergist) | ACTH (from POMC) | Adrenal cortex (zona fasciculata) | Cortisol |
| HPT | TRH | TSH (thyrotropin) | Thyroid follicle | T3 / T4 |
| HPG | GnRH (pulsatile) | LH, FSH | Gonads (testis / ovary) | Testosterone / oestrogen (+ inhibin) |
| Somatotropic | GHRH (+ somatostatin inhibitor) | Growth hormone (GH) | Liver → IGF-1 | IGF-1 (+ GH direct) |
| Prolactin | None required (PRF minor) | Prolactin | Breast | Dopamine (TONIC inhibition — no negative feedback from product) |
Three structural features recur across the axes and are favourite exam points: [1]
- Negative feedback is the rule — the end-product of each axis inhibits both the hypothalamus and the anterior pituitary. This is why exogenous glucocorticoids suppress the HPA axis (and why abrupt withdrawal causes adrenal crisis), and why exogenous thyroid hormone suppresses TSH.
- Prolactin is the exception — it is under TONIC DOPAMINERGIC INHIBITION rather than product-mediated negative feedback. Dopamine antagonists (antipsychotics, metoclopramide) raise prolactin; dopamine agonists (cabergoline, bromocriptine) lower it. There is no recognised hypothalamic prolactin-releasing hormone of equivalent importance.
- Pulsatility matters — GnRH must be delivered in pulses to maintain gonadotroph responsiveness; a continuous GnRH analogue (leuprorelin, goserelin) paradoxically SUPPRESSES the gonadal axis by downregulating gonadotroph receptors. This underlies medical androgen-deprivation therapy in prostate cancer. [1]
The generic three-tier negative-feedback loop
- A sensory or metabolic input (circadian rhythm, stress, plasma osmolality, substrate level) is detected by hypothalamic neurons
- Hypothalamic neurons secrete a releasing hormone into the hypophyseal portal system at the median eminence
- The releasing hormone binds G-protein-coupled receptors on specific anterior pituitary trophic cells, triggering hormone synthesis and release via second messengers (cAMP, IP3/Ca2+)
- The pituitary trophic hormone travels in the systemic circulation to its end-organ gland, where it stimulates synthesis and release of the end-product hormone
- The end-product acts on target tissues AND exerts negative feedback on BOTH the hypothalamus (releasing hormone) and the anterior pituitary (trophic hormone), closing the loop
- Any break in the loop (hypothalamic, pituitary, or end-organ failure) produces a recognisable hormone pattern: low end-product with HIGH trophic hormone = primary (end-organ) failure; low end-product with LOW trophic hormone = secondary (central) failure
Thyroid axis — TRH → TSH → T3/T4
The thyroid follicle is the structural unit: a sphere of follicular cells surrounding a central colloid lake filled with thyroglobulin. Thyroid hormone synthesis is unique in endocrinology because it couples an inorganic substrate (iodide) to an organic backbone (tyrosine) on a stored protein, and the whole process is rate-limited by iodide availability. [1]
Thyroid hormone synthesis — step by step
- Iodide uptake: the basal membrane of the follicular cell expresses the sodium-iodide symporter (NIS), which concentrates iodide 20-40x against its gradient using the sodium gradient maintained by Na/K-ATPase. (NIS is the target of radioactive iodine ablation; perchlorate blocks it.)
- Transport to colloid: iodide diffuses across the cell and is exported into the colloid at the apical membrane by pendrin and other anion transporters. (Pendrin defects cause Pendred syndrome — goitre with deafness.)
- Organification: at the apical-colloid interface, thyroid peroxidase (TPO) oxidises iodide to iodine (using H2O2 from DUOX2) and iodinates tyrosyl residues on thyroglobulin → monoiodotyrosine (MIT) and diiodotyrosine (DIT). (TPO is the target of carbimazole/propylthiouracil; H2O2 generation is also the target.)
- Coupling: TPO couples one DIT + one MIT to form T3 (triiodothyronine), and two DIT to form T4 (thyroxine). Coupled hormone remains stored on thyroglobulin in the colloid — weeks of reserve.
- Endocytosis and release: TSH stimulates endocytosis of colloid thyroglobulin back into the cell, fusion with lysosomes, proteolysis, and release of T4 and T3 into the bloodstream. Uncoupled MIT/DIT are deiodinated and the iodide recycled.
- Peripheral conversion: secreted T4 (about 80% of output) is converted in liver, kidney and muscle by 5'-deiodinase enzymes (D1, D2) to the active T3; an alternative pathway (D3) generates inactive reverse T3.
The single most important concept is that the thyroid secretes mainly T4 — a prohormone — and that ~80% of circulating active T3 is generated peripherally by deiodination. T3 is roughly 4-5 times more biologically active than T4 and is the principal ligand for the nuclear thyroid hormone receptor, which acts as a ligand-gated transcription factor controlling basal metabolic rate, thermogenesis, Na/K-ATPase activity, cardiac beta-adrenergic receptor density and skeletal growth.[2]
The three iodothyronine deiodinases
| Enzyme | Location | Reaction | Role / clinical relevance |
|---|---|---|---|
| D1 (5') | Liver, kidney, thyroid | T4 → T3 (and rT3 → T2) | Major source of plasma T3; inhibited by cytokines, propylthiouracil, amiodarone, contrast |
| D2 (5') | Brain, pituitary, skeletal muscle, brown fat | T4 → T3 | Local intracellular T3 for brain/pituitary feedback; preserved in illness |
| D3 (5) | Placenta, fetus, skin; INDUCED in critical illness | T4 → rT3; T3 → T2 | The INACTIVATING pathway — shunts hormone away from active T3 |
Adrenal axis — CRH → ACTH → cortisol
The adrenal cortex has three concentric zones, remembered by "GFR — salt, sugar, sex; the deeper you go, the sweeter it gets": the outer zona glomerulosa makes the mineralocorticoid aldosterone (salt); the middle zona fasciculata makes the glucocorticoid cortisol (sugar); the inner zona reticularis makes the androgens DHEA and androstenedione (sex). The medulla, derived from neural crest, is a modified sympathetic ganglion that synthesises and stores catecholamines.[1]
HPA axis activation and cortisol synthesis — step by step
- Stimulus: the paraventricular nucleus receives input from circadian rhythm, pain, hypoglycaemia, hypovolaemia, cytokines (IL-1, IL-6, TNF-alpha) and emotional stress
- CRH release into the hypophyseal portal system (ADH from the same nucleus synergises with CRH to amplify ACTH release)
- ACTH secretion from corticotroph cells of the anterior pituitary — cleaved from pro-opiomelanocortin (POMC), which also yields beta-lipotrophin and beta-endorphin
- ACTH binds melanocortin-2 receptors on zona fasciculata cells → Gs/cAMP/PKA → StAR protein shuttles cholesterol into mitochondria
- Steroidogenesis: cholesterol → pregnenolone (rate-limiting step, catalysed by cholesterol side-chain cleavage enzyme CYP11A1) → sequential steps (3beta-HSD, 17alpha-hydroxylase, 21-hydroxylase, 11beta-hydroxylase) → cortisol
- Release and transport: cortisol enters blood largely bound to cortisol-binding globulin (CBG) and albumin; only the FREE fraction is active. In inflammation CBG falls, so free cortisol rises disproportionately to total
- Cellular action: cortisol crosses the cell membrane, binds the intracellular glucocorticoid receptor, translocates to the nucleus and binds glucocorticoid response elements (GREs) — genomic effects take HOURS (and some rapid non-genomic effects via membrane receptors)
- Negative feedback: cortisol suppresses CRH and ACTH — partly overridden in critical illness by sustained cytokine and neural input
Cortisol — the four essential effects in critical illness
| Effect | Mechanism | Consequence if deficient |
|---|---|---|
| Permissive vascular tone | Upregulates alpha-1 adrenergic receptor expression and post-receptor signalling, permitting catecholamine vasoconstriction | Vasoplegic, catecholamine-resistant shock |
| Anti-inflammatory / immunosuppressive | Inhibits NF-kB, phospholipase A2 (reduces prostaglandins/leukotrienes), pro-inflammatory cytokines; induces IL-10, annexin-1 | Uncontrolled, exaggerated inflammation |
| Metabolic — gluconeogenesis | Induces PEPCK and glucose-6-phosphatase; mobilises amino acids (muscle) and glycerol (fat); opposes insulin | Inability to generate glucose — fasting hypoglycaemia |
| Volume / electrolyte (mild mineralocorticoid) | Weak mineralocorticoid receptor binding; at high levels overwhelms renal 11beta-HSD2 | Sodium loss, hyperkalaemia (only prominent in absolute adrenal failure) |
The catecholamines complete the adrenal stress response. Preganglionic sympathetic fibres synapse on chromaffin cells of the adrenal medulla, which release a ~4:1 mixture of adrenaline and noradrenaline (and a small amount of dopamine) into the bloodstream. Tyrosine → DOPA (tyrosine hydroxylase, rate-limiting) → dopamine → noradrenaline → adrenaline (catalysed by PNMT, induced by cortisol — one reason the cortex and medulla sit together). Catecholamines act within seconds via G-protein-coupled adrenergic receptors: beta-1 (heart — chronotropy, inotropy, renin release), beta-2 (bronchodilation, vasodilation, gluconeogenesis, insulin/glucagon modulation), alpha-1 (vasoconstriction) and alpha-2 (presynaptic autoregulation, inhibition of insulin release).[5]
Stress response — the integrated endocrine reaction to critical illness
Critical illness triggers a coordinated neuroendocrine cascade whose purpose is to maintain perfusion, circulating volume and substrate (glucose, free fatty acids) delivery to vital organs while the body fights, bleeds or heals. The response has two temporal phases. The acute phase (hours to days) is dominated by the sympathetic nervous system and the hypothalamic-pituitary axes: catecholamines, cortisol, ADH, aldosterone and glucagon all rise. This phase is CATABOLIC by design — glycogen, fat and protein are broken down to fuel the response. The chronic phase (weeks), seen in prolonged ICU stay, becomes maladaptive: anterior pituitary secretion becomes pulsatile and disordered, peripheral hormone activation falls (low T3, low IGF-1), and sustained catabolism produces ICU-acquired weakness, hyperglycaemia and immunosuppression.[5][9]
The hormonal players divide into three functional groups: [1]
- Haemodynamic / volume-maintaining: cortisol (permissive for catecholamines), catecholamines, ADH/vasopressin, and the RAAS (angiotensin II, aldosterone).
- Substrate-mobilising (counter-regulatory): cortisol, glucagon, catecholamines, growth hormone — all OPPOSE insulin to raise glucose and free fatty acids.
- Immune-modulating: cortisol (anti-inflammatory) balanced against pro-inflammatory cytokines (IL-1, IL-6, TNF-alpha), which both drive the stress response and induce hormone resistance. [1]
Acute vs chronic phases of the endocrine stress response
| Feature | Acute phase (hours-days) | Chronic phase (weeks) |
|---|---|---|
| Cortisol | Markedly elevated (600-1500 nmol/L) | Falls toward normal or low; pulsatility lost |
| Catecholamines | High | Normalise |
| ADH / aldosterone | High (water and sodium retained) | Persist; contributes to positive fluid balance |
| Thyroid | T3 falls (sick euthyroid), rT3 rises | Low T3, may develop low T4 |
| Growth hormone / IGF-1 | GH rises, IGF-1 falls (resistance) | Both low (catabolic dominance) |
| Gonadal | Testosterone falls acutely | Remains suppressed |
| Net metabolic state | Catabolic hypermetabolism | Persistent catabolism, weakness, hyperglycaemia |
Critical illness-related corticosteroid insufficiency (CIRCI)
CIRCI — the term coined by the 2008 international consensus task force — is the HPA axis dysfunction that develops DURING critical illness.[1] It differs from classical (Addisonian) adrenal insufficiency in three ways: (1) it is relative — cortisol production is inadequate FOR THE DEGREE OF STRESS, not absolutely absent; (2) it combines adrenal insufficiency with tissue glucocorticoid resistance (cytokines downregulate and impair the glucocorticoid receptor, so even normal cortisol concentrations are functionally inadequate); and (3) it is transient, resolving as the critical illness resolves.[5]
CIRCI vs primary (Addisonian) adrenal insufficiency
| Feature | CIRCI (critical illness) | Primary adrenal insufficiency |
|---|---|---|
| Nature of deficit | Relative + tissue resistance | Absolute — cortisol absent/minimal |
| ACTH | Low / normal / high (variable) | Markedly HIGH (loss of feedback) |
| Aldosterone | Preserved (RAAS intact) | Deficient (zona glomerulosa destroyed) |
| Electrolytes | Often normal | Hyponatraemia + HYPERKALAEMIA |
| Pigmentation | Absent | Present (high ACTH/MSH) |
| Onset | Develops during critical illness | Pre-existing, decompensated by stress |
| Diagnosis | Clinical (vasopressor-refractory shock) | Cortisol + ACTH (250 mcg) stimulation test |
| Treatment | Hydrocortisone 200 mg/day, then wean | Lifelong hydrocortisone + fludrocortisone |
When to suspect CIRCI: a patient in septic shock who requires escalating or high-dose vasopressors (noradrenaline over about 0.25 mcg/kg/min) despite adequate fluid resuscitation. The 2008 consensus and the Surviving Sepsis Campaign do NOT recommend a random cortisol level or an ACTH stimulation test to DECIDE whether to treat septic shock — the diagnosis is CLINICAL and treatment is empirical hydrocortisone 200 mg/day (50 mg IV 6-hourly or continuous infusion). The classic thresholds (random cortisol under 276 nmol/L, or delta cortisol under 250 nmol/L after 250 mcg ACTH) remain worth knowing as physiology but are not used as treatment gates. Dexamethasone is NOT recommended — it lacks mineralocorticoid activity and suppresses the axis; hydrocortisone must be WEANED, never stopped abruptly, to avoid rebound shock.[1]
Glucose homeostasis — insulin, glucagon and the counter-regulatory hormones
The glucose homeostasis
- The insulin (the beta cell; the - the - the).[1]
- The glucagon (the alpha cell; the - the - the).[1]
- The counter-regulatory (the cortisol, the adrenaline, the GH).[1]
Blood glucose is held within a narrow window (about 4-7 mmol/L) by the balance of insulin (the only hormone that LOWERS glucose) against a redundant set of counter-regulatory hormones that RAISE it (glucagon, adrenaline, noradrenaline, cortisol, growth hormone). The redundancy on the raising side reflects the evolutionary primacy of avoiding hypoglycaemia — the brain is obligately glucose-dependent and cannot tolerate fuel shortage, so multiple hormones back each other up. Insulin stands alone on the lowering side, which is why beta-cell failure (type 1 diabetes) and insulin resistance (type 2, stress) produce hyperglycaemia so readily. [1]
Insulin is synthesised in beta cells of the pancreatic islets as preproinsulin → proinsulin → insulin + C-peptide. Release is triggered by glucose entering beta cells via GLUT2, being phosphorylated by glucokinase (the beta-cell glucose sensor), and raising intracellular ATP, which closes ATP-sensitive K+ channels → depolarisation → voltage-gated calcium influx → exocytosis of insulin granules (the target of sulphonylureas, which directly close K-ATP). Insulin acts via the insulin receptor (receptor tyrosine kinase → IRS-1/PI3K/Akt) to: translocate GLUT4 to skeletal muscle and adipose membranes (glucose uptake); stimulate glycogen synthesis (glycogen synthase); stimulate lipogenesis and inhibit lipolysis; and stimulate protein synthesis. C-peptide is secreted in equimolar amounts with insulin and is the marker used to distinguish endogenous from exogenous insulin.[9]
Glucagon, from alpha cells, is the principal counter-regulatory hormone: it activates hepatic glycogen phosphorylase (via cAMP/PKA) to drive glycogenolysis and induces gluconeogenesis, raising glucose within minutes. The other counter-regulatory hormones — adrenaline, cortisol, growth hormone — act more slowly and on a longer time base but converge on the same hepatic gluconeogenic and glycogenolytic enzymes. They also all induce peripheral insulin resistance, ensuring glucose is diverted away from muscle/fat toward the brain.[9]
Pathogenesis of stress hyperglycaemia in critical illness
- Stress activates the sympathetic nervous system and HPA axis → cortisol, adrenaline, noradrenaline, glucagon and growth hormone all rise
- Glucagon drives hepatic glycogenolysis and gluconeogenesis
- Cortisol induces gluconeogenic enzymes (PEPCK, glucose-6-phosphatase) and mobilises gluconeogenic substrates (amino acids from muscle, glycerol from fat)
- Catecholamines stimulate hepatic glycogenolysis (beta-2), inhibit insulin release (alpha-2 on beta-cell) and stimulate glucagon (beta-2 on alpha-cell)
- Cytokines (IL-6, TNF-alpha) induce INSULIN RESISTANCE in skeletal muscle and adipose tissue (impaired IRS-1 / PI3K / GLUT4 signalling) — glucose cannot enter cells
- Net effect: hepatic glucose output exceeds peripheral uptake → HYPERGLYCAEMIA, even though insulin levels are HIGH (not low)
- Consequences: hyperosmolarity, osmotic diuresis (dehydration, electrolyte loss), impaired neutrophil function, oxidative stress — but also guaranteed glucose delivery to the glucose-dependent brain
The pivotal clinical lesson is the MANAGEMENT of stress hyperglycaemia. In 2001, van den Berghe's Leuven trial suggested that TIGHT glycaemic control (80-110 mg/dL; 4.4-6.1 mmol/L) dramatically reduced mortality in surgical ICU patients.[4] In 2009 the multinational NICE-SUGAR trial (6104 patients) REVERSED this conclusion: intensive control INCREASED 90-day mortality (27.5% vs 24.9%) driven by a 6-fold increase in severe hypoglycaemia (glucose under 2.2 mmol/L).[3] Modern practice targets 8-10 mmol/L and prioritises HYPOGLYCAEMIA AVOIDANCE, because a single severe hypoglycaemic event is independently associated with death.[6]
Sick euthyroid syndrome (nonthyroidal illness syndrome)
In critical illness the peripheral activation of thyroid hormone is REPROGRAMMED in what is called the sick euthyroid syndrome or nonthyroidal illness syndrome.[2] The pattern is an adaptive, energy-conserving down-tuning of thyroid hormone action:
- T3 FALLS (low T3 syndrome) — 5'-deiodinase activity (D1/D2) is suppressed by cytokines, cortisol and drugs, while the inactivating enzyme D3 is induced, SHUNTING T4 away from active T3 and toward inactive reverse T3.
- Reverse T3 RISES — the inactive metabolite accumulates.
- T4 is normal or low — in prolonged critical illness T4 also falls ("low T4 syndrome").
- TSH is normal or LOW — distinguishing it from primary hypothyroidism, where TSH is HIGH. [1]
This is an ADAPTIVE response: by lowering active T3, the body reduces basal metabolic rate, oxygen consumption and catabolism, conserving energy during the crisis. The clinical error to avoid is treating these abnormal thyroid function tests with levothyroxine — multiple studies show NO benefit (and potential harm). Levothyroxine is indicated ONLY if intrinsic thyroid disease coexists (known Hashimoto's, post-thyroidectomy, amiodarone-induced destructive thyroiditis), recognised by a genuinely ELEVATED TSH.[2]
Interpreting thyroid function tests in the ICU
| Scenario | TSH | Free T4 | Free T3 | rT3 | Interpretation |
|---|---|---|---|---|---|
| Sick euthyroid (early) | Normal/low | Normal | LOW | HIGH | Adaptive — do NOT treat |
| Sick euthyroid (prolonged) | Low | LOW | Low | High | Advanced — still do NOT treat |
| Primary hypothyroidism | HIGH | Low | Low/normal | Low | Treat with levothyroxine |
| Secondary (pituitary) hypothyroidism | Low/normal | Low | Low | — | Treat; co-exists with other pituitary failure |
| Dopamine infusion | Low | Low | Low | — | Dopamine SUPPRESSES TSH — artefact; recheck off dopamine |
| Amiodarone | Variable | Variable | Variable | — | Both hypo- and hyperthyroidism possible — check TSH + T4 |
Vasopressin physiology and deficiency in shock
Antidiuretic hormone (ADH, also arginine vasopressin) is synthesised in the supraoptic and paraventricular nuclei of the hypothalamus and released from the posterior pituitary. It has two distinct regulatory inputs that compete for control of its release, and understanding which dominates is the key to ICU sodium and water physiology. [1]
- OSMOTIC regulation (the dominant day-to-day control): hypothalamic osmoreceptors detect plasma osmolality with extraordinary sensitivity. ADH release begins at about 280 mOsm/kg and rises steeply to about 295 mOsm/kg, at which point thirst is also triggered. ADH acts on V2 receptors in the renal collecting duct → inserts aquaporin-2 water channels → water reabsorption → concentrates urine and dilutes plasma back toward normal. A slightly high sodium (hypertonicity) is therefore the most powerful stimulus to ADH.
- BARORECEPTOR (non-osmotic) regulation: carotid sinus and aortic arch baroreceptors sense arterial stretch. A FALL in blood pressure or effective circulating volume (over about 10%) is a powerful stimulus to ADH release EVEN IF plasma osmolality is low. This is the "non-osmotic ADH release" of shock, heart failure and cirrhosis: the body sacrifices osmolality to preserve circulating volume. ADH also acts on V1a receptors on vascular smooth muscle → vasoconstriction → a pressor effect exploited therapeutically in vasoplegic shock. [1]
ADH release — two competing pathways
- Osmotic pathway: plasma osmolality rises (over 280 mOsm/kg) → osmoreceptors shrink → signal to hypothalamus → ADH release → V2 receptors → aquaporin-2 insertion → water retained → osmolality normalised
- Baroreceptor pathway: arterial pressure/volume drops (over 10%) → carotid sinus/aortic arch unloading → vagal and glossopharyngeal afferents to brainstem → hypothalamus → ADH release → V1a vasoconstriction (pressor) + V2 water retention → BP and volume restored
- In health: the osmotic pathway dominates — ADH adjusts minute-to-minute to sodium intake and water availability
- In critical illness (shock, sepsis, post-op, nausea, pain, mechanical ventilation): baroreceptor and other non-osmotic inputs OVERRIDE the osmotic pathway → ADH is high EVEN WHEN osmolality is low → water retained in excess of sodium → DILUTIONAL HYPONATRAEMIA (the SIADH pattern)
The three vasopressin receptors
| Receptor | Location | G-protein coupling | Effect |
|---|---|---|---|
| V1a | Vascular smooth muscle, platelets, liver | Gq / IP3 / Ca2+ | Vasoconstriction, platelet aggregation, glycogenolysis |
| V1b (V3) | Anterior pituitary (corticotrophs) | Gq / IP3 / Ca2+ | ACTH release (synergy with CRH) |
| V2 | Renal collecting duct (principal cells) | Gs / cAMP / PKA | Aquaporin-2 insertion → water reabsorption |
Sodium and water balance — ADH, V2 receptors and the RAAS
Sodium and water are regulated in tandem but by partly separable systems. Water balance is governed by ADH (V2/aquaporin-2, described above) and thirst. Sodium balance is governed primarily by the renin-angiotensin-aldosterone system (RAAS) and by atrial natriuretic peptide. In health, plasma sodium reflects water balance (a sodium abnormality is almost always a water problem), while total body sodium (volume status) reflects sodium balance — a distinction central to interpreting ICU electrolytes.[1]
RAAS activation in shock
- Reduced renal perfusion / low pressure at the afferent arteriole / low luminal sodium at the macula densa activates the juxtaglomerular apparatus
- Renin is released from juxtaglomerular cells into the circulation
- Renin cleaves circulating angiotensinogen (from the liver) to angiotensin I
- Angiotensin-converting enzyme (ACE), predominantly in pulmonary vascular endothelium, converts angiotensin I to angiotensin II
- Angiotensin II (one of the most potent vasoconstrictors known, via AT1 receptors) constricts arterioles, preferentially the efferent arteriole of the glomerulus (preserving GFR), stimulates the adrenal zona glomerulosa to release aldosterone, stimulates ADH and thirst, and enhances sympathetic tone
- Aldosterone acts on distal convoluted tubule and collecting duct principal cells (via mineralocorticoid receptor → ENaC sodium channel and Na/K-ATPase) to reabsorb sodium (and water) in exchange for potassium and hydrogen ion excretion → expands circulating volume and supports blood pressure
- The system is appropriately intense in any state of perceived volume loss (shock, haemorrhage, dehydration, heart failure, sepsis) and sustains BP and glomerular perfusion when sympathetic tone alone is insufficient
The clinical relevance of RAAS activation in critical illness is threefold. First, RAAS blockers (ACE inhibitors, ARBs) can precipitate or worsen shock by removing this defence (loss of angiotensin-II vasoconstriction, loss of efferent arteriolar tone); they should generally be held in acute critical illness. Second, aldosterone-driven potassium excretion explains the hypokalaemia often seen in resuscitated patients. Third, sustained sodium and water retention contributes to the positive fluid balance and oedema of prolonged critical illness — a rationale for later active deresuscitation.[5]
The interaction between ADH and RAAS is the explanation for the near-universal hyponatraemia of the sick ICU patient. When non-osmotic ADH release is APPROPRIATE (true hypovolaemia, shock), the hyponatraemia is a correct physiological response — treat the underlying volume deficit with isotonic saline. When non-osmotic ADH release is INAPPROPRIATE (euvolaemic, as in SIADH from pneumonia, subarachnoid haemorrhage, malignancy, or drugs), the retained water must be managed with fluid restriction, and corrected SLOWLY (no more than 8-10 mmol/L in 24 h) to avoid osmotic demyelination syndrome.[5]
Calcium homeostasis — PTH, vitamin D and calcitonin
The calcium homeostasis
- The PTH (the parathyroid; the - the - the).[1]
- The vitamin D (the - the - the).[1]
- The calcitonin (the thyroid the C cell; the - the - the).[1]
Plasma calcium is maintained in a narrow range (ionised calcium about 1.1-1.3 mmol/L) by three hormones acting on three organs (gut, kidney, bone). About 50% of plasma calcium is ionised (biologically active), 40% is protein-bound (mostly albumin), and 10% is complexed to anions — which is why the corrected calcium formula adds 0.02 mmol/L per g/L of albumin below 40 g/L. Acidosis increases the ionised fraction (displaces calcium from albumin); alkalosis decreases it.[8]
Parathyroid hormone (PTH), from the chief cells of the four parathyroid glands, is the minute-to-minute regulator. It is released when the calcium-sensing receptor (CaSR) on chief cells detects a FALL in ionised calcium, and it RAISES calcium by: (1) BONE — stimulating osteoclasts (indirectly, via RANKL/osteoprotegerin) to resorb bone and release calcium and phosphate; (2) KIDNEY — increasing calcium reabsorption in the distal convoluted tubule (via apical TRPV5 and basolateral Na/Ca exchange) while DECREASING phosphate reabsorption in the proximal tubule (phosphaturia — important because phosphate binds calcium and because high phosphate suppresses 1-alpha-hydroxylase); and (3) GUT — indirectly, by stimulating renal 1-alpha-hydroxylase to activate vitamin D. [1]
Vitamin D (cholecalciferol) is the slower, day-to-day regulator. Synthesised in the skin from 7-dehydrocholesterol under UV-B light (and ingested in the diet), it undergoes 25-hydroxylation in the liver (CYP2R1) to calcifediol (25-OH-D, the storage form measured in blood) and then 1-alpha-hydroxylation in the kidney (CYP27B1, stimulated by PTH and hypophosphataemia, inhibited by FGF-23 and hyperphosphataemia) to the active calcitriol (1,25-(OH)2-D). Calcitriol acts via the vitamin D receptor (nuclear) to increase intestinal absorption of both calcium AND phosphate (by inducing TRPV6 calcium channel and calbindin), and it synergises with PTH on bone.[8]
Calcitonin, from the parafollicular C cells of the thyroid, is the minor, opposing hormone: it LOWERS calcium by inhibiting osteoclast activity. Its physiological role in humans is small (thyroidectomy does not cause hypercalcaemia, and calcitonin excess from medullary thyroid cancer causes only modest hypocalcaemia), but it is therapeutically useful (Paget disease, hypercalcaemia of malignancy).[1]
The calcium homeostatic response to hypocalcaemia
- Ionised calcium FALLS (e.g. acute pancreatitis saponification, massive transfusion citrate, sepsis, renal failure)
- The calcium-sensing receptor (CaSR) on parathyroid chief cells is unloaded → PTH release within seconds
- PTH acts on BONE within minutes → osteoclast activation → calcium and phosphate released into plasma
- PTH acts on KIDNEY → increased distal calcium reabsorption AND decreased proximal phosphate reabsorption (phosphaturia) — the phosphaturia is essential to prevent the resorbed phosphate from re-precipitating calcium
- PTH stimulates renal 1-alpha-hydroxylase → more calcitriol → increased GUT calcium (and phosphate) absorption over hours to days
- Rising calcitonin (minor, from C cells) opposes any overshoot by inhibiting osteoclasts
- Net effect: plasma calcium restored to normal; if the stimulus persists (chronic renal failure, vitamin D deficiency), the system runs persistently high (secondary hyperparathyroidism) with elevated PTH
The three calcium-regulating hormones at a glance
| Hormone | Source | Trigger | Effect on Ca2+ | Effect on PO4 | Effect on bone | Time-frame |
|---|---|---|---|---|---|---|
| PTH | Parathyroid chief cells | Low ionised Ca2+ (via CaSR) | RAISES | LOWERS (phosphaturia) | Resorption | Seconds-minutes |
| Calcitriol (1,25-(OH)2-D) | Kidney (1alpha-hydroxylase) | PTH, low PO4; blocked by high PO4/FGF-23 | RAISES | RAISES | Mineralisation | Hours-days |
| Calcitonin | Thyroid C cells | High Ca2+ | LOWERS | LOWERS | Inhibits resorption | Minutes (minor role) |
Exam-style short-answer questions
SAQ — Stress hyperglycaemia in septic shock
10 minutes · 10 marks
A 68-year-old man with no prior diabetes is admitted to ICU with severe sepsis from a perforated sigmoid diverticulum. After source control and 30 mL/kg crystalloid he remains vasopressor-dependent. On day 2 his glucose is 16.4 mmol/L, lactate 3.2 mmol/L, Na 134, K 4.8; he is on no insulin. The bedside nurse asks whether to start an insulin infusion and what glucose target to aim for.
SAQ — Perioperative HPA-axis suppression in chronic steroid therapy
10 minutes · 10 marks
A 58-year-old woman with severe rheumatoid arthritis has taken oral prednisolone 15 mg daily for 6 years. She is admitted for elective total hip replacement and the surgical team has asked ICU to advise on perioperative glucocorticoid management. She has mild central adiposity but no overt Cushingoid features. A preoperative 09:00 cortisol is 95 nmol/L.
Clinical pearls
Red flags
Key trials and evidence
Marik 2008 — CIRCI consensus (PMID 18496365)
Source
Critical Care Medicine 2008 — international task force, American College of Critical Care Medicine
Key contribution
Coined the term CRITICAL ILLNESS-RELATED CORTICOSTEROID INSUFFICIENCY (CIRCI) — defined as adrenal insufficiency PLUS tissue glucocorticoid resistance during critical illness
Key recommendation
Hydrocortisone 200 mg/day (50 mg q6h or continuous) for vasopressor-dependent septic shock — WITHOUT an ACTH stimulation test. Dexamethasone NOT recommended. Wean, never stop abruptly
Diagnostic threshold (for knowledge)
Random cortisol under 276 nmol/L (10 mcg/dL) OR delta cortisol under 250 nmol/L (9 mcg/dL) after 250 mcg ACTH — but treatment in sepsis is CLINICAL, not test-based
Clinical bottom line
The foundational document for ICU adrenal physiology practice — treat the SHOCK, not the number
NICE-SUGAR 2009 — intensive vs conventional glucose control (PMID 19318384)
Source
New England Journal of Medicine 2009 — multinational RCT, 6104 critically ill adults (medical + surgical ICU)
Intervention
Intensive glucose control 4.5-6.0 mmol/L (81-108 mg/dL) vs conventional target 10.0 mmol/L or less (180 mg/dL)
Primary outcome
90-day all-cause mortality: INTENSIVE 27.5% vs CONVENTIONAL 24.9% (OR 1.14, P=0.02) — intensive control INCREASED mortality
Harm
Severe hypoglycaemia (glucose 2.2 mmol/L or less): 6.8% intensive vs 0.5% conventional (P under 0.001) — the mechanism of harm
Clinical bottom line
Ended the tight-glycaemic-control era; established the modern target of 8-10 mmol/L with hypoglycaemia avoidance as the priority
Russell 2008 — VASST, vasopressin vs norepinephrine in septic shock (PMID 18305265)
Source
New England Journal of Medicine 2008 — multicentre RCT, 778 patients with septic shock on vasopressors
Intervention
Low-dose vasopressin 0.01-0.04 units/min added to open-label norepinephrine, vs norepinephrine alone
Primary outcome
28-day mortality similar (vasopressin 35.4% vs norepinephrine 39.3%) — vasopressin NON-INFERIOR
Subgroup signal
In LESS severe septic shock (norepinephrine under 15 mcg/min at baseline), vasopressin reduced 28-day mortality (26.5% vs 35.7%); no benefit in more severe shock
Clinical bottom line
Justifies low-dose vasopressin as a catecholamine-sparing ADD-ON to norepinephrine in septic shock; never first-line, never above 0.04 units/min
van den Berghe 2001 — intensive insulin in surgical ICU (PMID 11794168)
Source
New England Journal of Medicine 2001 — single-centre RCT, 1548 surgical ICU patients (predominantly cardiac surgery)
Intervention
Intensive insulin to maintain glucose 4.4-6.1 mmol/L (80-110 mg/dL) vs conventional 10.0-11.1 mmol/L (180-200 mg/dL)
Primary outcome
ICU mortality reduced 8.0% to 4.6%; benefit concentrated in patients staying more than 5 days (20.2% to 10.6%); also reduced bacteraemia, AKI, polyneuropathy, transfusions
Why it was superseded
Single-centre, surgical-only population; a later medical-ICU trial (2006) showed equivocal results and excess hypoglycaemia; NICE-SUGAR (2009) refuted the mortality benefit in a mixed population
Clinical bottom line
Historically pivotal — launched tight glycaemic control — but no longer the basis for modern targets; essential to know for exam comparison with NICE-SUGAR
Cooper & Stewart 2003 — corticosteroid insufficiency in acutely ill (PMID 12594318)
Source
New England Journal of Medicine 2003 — review
Key contribution
Synthesised the evidence that critically ill patients develop RELATIVE adrenal insufficiency: cortisol is inappropriately low for the degree of stress, CBG falls (free cortisol rises), and tissue glucocorticoid resistance emerges
Clinical relevance
Provided the conceptual foundation for empirical hydrocortisone in vasopressor-refractory septic shock and for why random total cortisol is misleading
Clinical bottom line
The classic reference for the permissive vascular effect of cortisol and the pitfalls of biochemical adrenal testing in the critically ill
Prognosis
The endocrine axes in critical illness behave as biomarkers of severity as much as therapeutic targets. Stress hyperglycaemia: both the mean glucose and its variability, and crucially any episode of hypoglycaemia, are independent predictors of mortality — glycaemic control quality is a measurable marker of ICU performance.[6][3] CIRCI: the need for hydrocortisone in septic shock identifies a sicker subgroup, but within that group hydrocortisone accelerates shock reversal and is catecholamine-sparing; mortality benefit is modest and confined to the most severe (vasopressor-refractory) cases.[1] Sick euthyroid syndrome: the depth and persistence of the low-T3/low-T4 pattern parallel illness severity — a falling T4 is a poor prognostic marker — but it remains an adaptive response that does not benefit from thyroid hormone replacement.[2] Relative vasopressin deficiency: identifying and treating it with low-dose vasopressin is catecholamine-sparing and may benefit less-severe septic shock (VASST subgroup), but it is not a mortality-changing monotherapy.[7] Across all axes, the overarching principle is to SUPPORT the endocrine response where it fails (hydrocortisone in CIRCI, vasopressin in vasoplegic shock, controlled insulin for harmful hyperglycaemia) and to RESIST over-treating the adaptive ones (sick euthyroidism, modest stress hyperglycaemia) — the ICU endocrine skill is judgement, not reflex.[9]
References
- [1]Marik PE, Pastores SM, Annane D, et al Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine Crit Care Med, 2008.PMID 18496365
- [2]Adler SM, Wartofsky L The nonthyroidal illness syndrome Endocrinol Metab Clin North Am, 2007.PMID 17673123
- [3]NICE-SUGAR Study Investigators; Finfer S, Chittock DR, Su SY, et al Intensive versus conventional glucose control in critically ill patients N Engl J Med, 2009.PMID 19318384
- [4]van den Berghe G, Wouters P, Weekers F, et al Intensive insulin therapy in critically ill patients N Engl J Med, 2001.PMID 11794168
- [5]Cooper MS, Stewart PM Corticosteroid insufficiency in acutely ill patients N Engl J Med, 2003.PMID 12594318
- [6]Krinsley JS, Egi M, Kiss A, et al Diabetic status and the relation of the three domains of glycemic control to mortality in critically ill patients: an international multicenter cohort study Crit Care, 2013.PMID 23452622
- [7]Russell JA, Walley KR, Singer J, et al Vasopressin versus norepinephrine infusion in patients with septic shock N Engl J Med, 2008.PMID 18305265
- [8]Holick MF Vitamin D deficiency N Engl J Med, 2007.PMID 17634462
- [9]Dungan KM, Braithwaite SS, Preiser JC Stress hyperglycaemia Lancet, 2009.PMID 19465235