ICU · Physiology / GI hepatic
GI & Hepatic Physiology
Also known as GI physiology · Hepatic physiology · Bilirubin metabolism · Urea cycle · Splanchnic circulation · Gut barrier · Enterohepatic circulation · Bile metabolism · Gastric acid secretion · Migrating motor complex · Liver regeneration · Hepatic drug metabolism
GI and hepatic physiology: the GI (the digestion, the absorption, the motility, the secretion; the gut barrier; the splanchnic circulation). The hepatic (the bilirubin metabolism — the heme to the biliverdin to the bilirubin; the conjugation; the enterohepatic circulation; the urea cycle; the glucose homeostasis; the coagulation factors; the detoxification; the bile). The clinical (the jaundice, the encephalopathy, the coagulopathy, the portal hypertension).
On this page & tools
Your progress
Saved locally on this device.
8 MCQs with explanations
Target exams
Overview & definition
The GI and hepatic physiology — the digestion, the absorption, the motility, the secretion; the liver (the bilirubin, the urea, the glucose, the coagulation, the detoxification, the bile). The splanchnic circulation. The gut barrier.[1]
GI physiology
- The digestion — the carbohydrates (the amylase), the proteins (the pepsin, the trypsin), the fats (the lipase, the bile salts).[1]
- The absorption — the small intestine (the - the - the - the).[1]
- The motility — the peristalsis; the migrating motor complex (the MMC — the fasting).[1]
- The secretion — the gastric acid (the parietal the cell; the H/K ATPase), the pancreatic (the bicarbonate, the enzymes), the bile.[1]
- The gut barrier — the mucosal; the tight junctions; the - the - the - the.[1]
- The splanchnic circulation — the dual supply (the hepatic artery 25 per cent, the portal vein 75 per cent).[1]
Swallowing reflex — the oropharyngeal to oesophageal sequence
Deglutition (swallowing) is a coordinated reflex that transfers a bolus from the mouth to the stomach while protecting the airway. It has three phases, only the first being truly voluntary: [1]
The three phases of swallowing
- ORAL PHASE (voluntary) — mastication and bolus formation; the tongue (XII/hypoglossal) pushes the bolus up and back against the hard palate toward the fauces. Duration voluntary, the only phase under conscious control.
- PHARYNGEAL PHASE (involuntary reflex, ~1 second) — the bolus touching the pharyngeal wall (sensory via IX glossopharyngeal and V trigeminal) triggers the swallowing centre in the medulla (nucleus ambiguus + tractus solitarius). The reflex simultaneously and irreversibly: (a) elevates the soft palate (X vagus) to close the nasopharynx (prevents nasal regurgitation); (b) presses the epiglottis down and tilts it over the larynx + approximates the vocal cords + elevates the larynx (prevents aspiration); (c) relaxes the upper oesophageal sphincter (UES, cricopharyngeus); (d) the pharyngeal constrictors strip the bolus into the oesophagus. Respiration is reflexly inhibited (deglutition apnoea) during this phase.
- OESOPHAGEAL PHASE (involuntary, ~8 seconds) — primary peristalsis (a peristaltic wave triggered by the swallowing centre, travelling by vagal reflex) pushes the bolus down; the lower oesophageal sphincter (LES) relaxes (via vagal inhibitory fibres releasing nitric oxide and VIP) to allow entry into the stomach. If residual bolus remains, secondary peristalsis (triggered by local distension via the intrinsic enteric reflex, no conscious sensation) clears it.
The sphincters — the UES (cricopharyngeus, striated muscle, baseline high pressure ~40-100 mmHg) and the LES (smooth muscle, ~10-30 mmHg) — are tonically CLOSED and relax only during swallowing (LES relaxation = transient lower oesophageal sphincter relaxation, TLESR, the mechanism of reflux). The crural diaphragm provides an external pinch on the LES (the anti-reflux barrier), which is why a hiatus hernia (loss of the diaphragmatic crural support) predisposes to gastro-oesophageal reflux.[1]
[1]Gastric acid secretion — the three phases and the H⁺/K⁺-ATPase
Gastric acid (HCl, pH ~1-2, up to ~2 L/day) is secreted by the parietal (oxyntic) cell of the gastric body and fundus. The final common effector is the H⁺/K⁺-ATPase (the proton pump) — an electroneutral ATP-driven exchanger on the apical (canalicular) membrane that pumps H⁺ into the lumen in exchange for K⁺. The proton pump is the target of proton pump inhibitors (PPIs, omeprazole) — irreversible (covalent) binders of the pump activated in the acidic canaliculus — and is the rationale for their near-complete acid suppression.[1]
Three secretagogues converge on the parietal cell: [1]
The three stimulants of parietal cell acid secretion
| Stimulant | Source | Receptor / mechanism | Notes |
|---|---|---|---|
| Histamine | ECL (enterochromaffin-like) cell in the gastric body (stimulated by gastrin via CCK-B / by vagal PACAP) | H₂ receptor (Gs / cAMP) on the parietal cell — the dominant paracrine amplifier | Target of H₂ blockers (ranitidine). ECL-cell hyperplasia/gastrinoma (Zollinger-Ellison) |
| Gastrin | G cell of the gastric antrum (and duodenum), released by peptides/amino acids, stomach distension, vagal GRP; inhibited by pH < 3 (negative feedback) | CCK-B (CCK-2) receptor on the ECL cell (and parietal cell) — acts MAINLY via histamine release from the ECL cell | The endocrine arm; measured as serum gastrin. Hypergastrinaemia from PPIs (loss of acid negative feedback) |
| Acetylcholine | Postganglionic parasympathetic (vagus) fibres from the enteric plexus | M₃ muscarinic receptor (Gq / IP₃-Ca²⁺) on the parietal cell (and on ECL and D cells) | Blocked by anticholinergics; the neurotransmitter arm of vagal stimulation |
Somatostatin (D cell of the gastric antrum and body, released when luminal pH < 3) is the master INHIBITOR — it suppresses gastrin (G cell), histamine (ECL cell) and the parietal cell directly (sst₂ receptor) — the negative-feedback brake that prevents acid over-secretion. This is why loss of the antrum (surgery) or achlorhydria (PPI/pernicious anaemia) causes hypergastrinaemia, and why somatostatin analogues (octreotide) reduce acid (and splanchnic) output.[2]
The three phases of gastric acid secretion
- CEPHALIC phase (~30% of response) — the sight, smell, taste and even thought of food activate higher cortical centres → the dorsal vagal complex → the vagus. Vagal output stimulates (a) the parietal cell directly (ACh → M₃), (b) the ECL cell (ACh + GRP → histamine), and (c) the G cell (GRP → gastrin). Demonstrable by sham feeding (chewing but not swallowing). Vagotomy abolishes the cephalic phase.
- GASTRIC phase (~60% of response, the largest) — food entering the stomach causes (a) distension (vagovagal and enteric reflex → parietal/G/ECL stimulation); (b) peptides and amino acids directly stimulate the G cell to release gastrin. The biggest contributor; the reason the bulk of acid is secreted when food is actually in the stomach.
- INTESTINAL phase (~10%) — chyme entering the duodenum: partially digested protein stimulates duodenal G cells to release gastrin; BUT simultaneously intestinal hormones (secretin, GIP, CCK, somatostatin) and the enterogastric reflex (acid/fat/hyperosmolarity in the duodenum → vagal and sympathetic → ↓ gastric motility and secretion) predominantly INHIBIT gastric acid — the enterogastric brake preventing acid overload of the duodenum.
The cellular activation cascade: stimulation → tubulovesicles (holding H⁺/K⁺-ATPase) fuse with the apical canalicular membrane → expansion of the secretory canaliculus → the proton pump inserts and secretes H⁺. At rest the pumps are sequestered intracellularly; with stimulation they translocate to the membrane — the basis for PPIs only inactivating ACTIVATED pumps (hence give PPIs ~30 min before a meal, when pumps are maximally active).[1]
Why acid is secreted — the physiological functions of gastric HCl
| Function | Mechanism / consequence when impaired |
|---|---|
| Activation of pepsinogen → pepsin | Acid (pH < 4) auto-cleaves pepsinogen (from chief cells) to pepsin; pepsin begins protein digestion (optimum pH ~1.8-3.5). Achlorhydria → impaired protein digestion |
| Antimicrobial barrier | pH ~1-2 kills most ingested bacteria. Achlorhydria (PPIs, pernicious anaemia, atrophic gastritis) → bacterial overgrowth, ↑ risk of enteric infection (C. difficile, Salmonella, Campylobacter) and SIBO |
| Iron and calcium absorption | Acid reduces ferric (Fe³⁺) → ferrous (Fe²⁺) and solubilises iron and calcium. PPIs → ↓ iron and calcium absorption (long-term fracture, hypochloraemia risk) |
| Stimulation of secretin | Acidic chyme (pH < 4.5) entering the duodenum releases secretin (S cells) → pancreatic bicarbonate — the acid-alkali "handover" |
Pancreatic enzyme secretion and activation — the zymogen safeguard
The exocrine pancreas secretes ~1.5 L/day of a bicarbonate-rich (pH ~8), enzyme-laden juice that is the principal digestive engine for carbohydrates, fats and proteins. Secretion is driven by two hormones: [1]
- Secretin (S cells of the duodenum, released by acidic chyme pH < 4.5) → duct cells secrete bicarbonate-rich, watery juice (neutralises acid, brings duodenal pH to ~6-7 for enzyme function).
- Cholecystokinin / CCK (I cells of the duodenum, released by amino acids, fatty acids, monoglycerides) → acinar cells secrete enzyme-rich juice; also contracts the gallbladder and relaxes the sphincter of Oddi.
- Vagal input (ACh, M₃) potentiates both (the cephalic and gastric phases of pancreatic secretion). [1]
Pancreatic enzyme activation — the safeguard against autodigestion
- Zymogen (proenzyme) synthesis — acinar cells synthesise digestive enzymes as INACTIVE precursors to avoid autodigesting the gland: trypsinogen, chymotrypsinogen, procarboxypeptidase, proelastase, prophospholipase, procolipase, plus amylase and lipase (secreted active) and pancreatic lipase (the rate-limiting fat-digesting enzyme).
- Enterokinase (enteropeptidase) — the master activator, a brush-border enzyme of the duodenal/jejunal mucosa — cleaves trypsinogen → trypsin. This is the critical, irreversible step that is deliberately kept OUTSIDE the pancreas (in the duodenal lumen).
- Trypsin autocatalysis — trypsin then activates all the other zymogens (chymotrypsinogen → chymotrypsin; procarboxypeptidase → carboxypeptidase; prophospholipase → phospholipase A₂; proelastase → elastase). Trypsin also autoactivates more trypsinogen (positive feedback).
- Intrapancreatic safeguards — (a) trypsin inhibitor (PSTI / SPINK1) co-secreted with the zymogens neutralises any trypsin prematurely activated inside the gland; (b) enzymes are packaged in zymogen granules and secreted by exocytosis only; (c) trypsin self-degradation and mesotrypsin degrade misfolded/trapped trypsin. Failure of these safeguards → acute pancreatitis (premature intracellular trypsinogen activation → autodigestion).[8]
The end-result is a suite of enzymes: amylase (starch → maltose/maltotriose/α-dextrins, secreted active), lipase (triglycerides → 2-monoglyceride + free fatty acids, requires colipase to function in the presence of bile salts), phospholipase A₂ (phospholipids → lysophospholipids + fatty acids), trypsin/chymotrypsin/carboxypeptidase/elastase (proteins → peptides and amino acids), and the nucleases (ribonuclease, deoxyribonuclease).[1]
Bile acid circulation — the enterohepatic loop
Bile has two functions — digestion (emulsification of dietary fat and micelle formation) and excretion (the route for conjugated bilirubin, cholesterol, and lipophilic xenobiotics). Hepatocytes synthesise the primary bile acids — cholic acid and chenodeoxycholic acid — from cholesterol (rate-limiting enzyme 7α-hydroxylase, CYP7A1; bile acid synthesis is the major pathway of cholesterol catabolism, and is feedback-inhibited by bile acids returning via the portal vein). They are conjugated with glycine or taurine (lowering their pKa so they remain soluble at intestinal pH as bile salts) → secreted across the canalicular membrane by BSEP (ABCB11) → bile flows to the gallbladder for storage and concentration → released post-prandially when CCK (released by I-cells in response to fat/amino acids) contracts the gallbladder and relaxes the sphincter of Oddi.[4]
In the duodenum, bile salts act as detergents: their amphipathic structure (hydrophobic steroid nucleus + hydrophilic hydroxyl/side-chain) emulsifies dietary triglycerides into small droplets, then forms mixed micelles (bile salts + fatty acids + monoglycerides + fat-soluble vitamins) that ferry lipids to the enterocyte brush border for absorption.[4]
The enterohepatic circulation of bile acids
- Secretion — conjugated bile acids are secreted into the bile canaliculus and stored in the gallbladder; released into the duodenum after a meal (CCK-driven).
- Lipid digestion — emulsification and micelle formation in the duodenum/jejunum; bile salts deliver fat digestion products to the enterocyte.
- Reabsorption (95% efficient) — bile acids are reabsorbed predominantly by ACTIVE transport in the TERMINAL ILEUM (apical sodium-dependent bile salt transporter, ASBT / SLC10A2) and passively throughout the gut. Only ~5% is lost in stool (0.5 g/day), the daily loss replaced by hepatic synthesis.
- Return to liver — reabsorbed bile acids return to the liver via the portal vein and are taken up by hepatocytes (NTCP at the sinusoid) → re-secreted into bile. The total pool (~3 g) recycles 6-10 times per day, so a 3 g pool delivers ~20-30 g/day of bile acids to the gut despite only ~0.5 g/day of new synthesis.
- Bacterial modification — colonic bacteria deconjugate (remove glycine/taurine) and dehydroxylate primary bile acids → secondary bile acids (deoxycholic, lithocholic), some of which are reabsorbed and contribute to the pool; lithocholic acid is relatively insoluble and mostly excreted.
The clinical corollaries: (1) terminal ileal disease/resection (Crohn's, surgical) disrupts reabsorption → bile acid loss in stool → depletion of the pool → fat malabsorption (steatorrhoea) and cholesterol gallstone formation (a depleted pool cannot keep cholesterol solubilised in bile); (2) bile acid malabsorption causes bile-salt diarrhoea (choleretic enteropathy, the colon irritated by secretory bile acids) — treat with a bile acid sequestrant (cholestyramine).[4][6]
Intestinal absorption — carbohydrates, proteins, fats, B₁₂, iron, calcium
The small intestine (surface area ~300 m² with villi and microvilli) is the principal absorptive organ. Each macronutrient class and several key micronutrients have specific transport mechanisms that are common exam material. [1]
Macronutrient digestion and absorption
| Nutrient | Digestion (enzymes) | Absorption (transporter / location) | Exit to blood |
|---|---|---|---|
| Carbohydrates | Salivary + pancreatic amylase → maltose, maltotriose, α-dextrins; brush-border disaccharidases (maltase, sucrase, lactase, α-dextrinase) → monosaccharides | Glucose & galactose: SGLT1 (Na⁺-dependent, secondary active, apical); GLUT2 (facilitated, basolateral). Fructose: GLUT5 (apical), GLUT2 (basolateral) — jejunum | Portal vein → liver |
| Proteins | Gastric pepsin; pancreatic trypsin/chymotrypsin/elastase/carboxypeptidase; brush-border peptidases → amino acids, di-/tri-peptides | Di-/tri-peptides: PepT1 (H⁺-coupled); free amino acids: several Na⁺-dependent AA transporters — jejunum | Portal vein |
| Fats (triglycerides) | Gastric + pancreatic lipase (colipase-dependent) → 2-monoglyceride + free fatty acids; bile-salt micelles deliver products to brush border | Diffusion across enterocyte membrane → re-esterified → packaged into chylomicrons → lymphatics (lacteals) — jejunum | LYMPH (NOT portal) via thoracic duct — the only macronutrient NOT going first to the liver |
The fat pathway is distinctive: because lipids are membrane-permeable, they diffuse into the enterocyte, are re-esterified in the smooth ER, packaged with apolipoproteins into chylomicrons, and secreted by exocytosis into the lymphatic lacteals (NOT the portal vein) — bypassing the hepatic first-pass. This is why medium-chain triglycerides (MCTs, C6-C12), which are water-soluble and absorbed directly into the portal vein (no micelle/chylomicron needed), are useful in fat malabsorption (pancreatic insufficiency, cholestasis, terminal ileal resection).[6]
Micronutrient absorption — the classic ICU-relevant examples
| Nutrient | Site | Mechanism | Clinical relevance |
|---|---|---|---|
| Vitamin B₁₂ (cobalamin) | Terminal ileum | Multi-step: (1) gastric R-protein (haptocorrin) binds B₁₂ in the acidic stomach; (2) pancreatic enzymes degrade R-protein in the duodenum; (3) B₁₂ binds intrinsic factor (IF) from gastric parietal cells; (4) IF-B₁₂ complex binds cubilin/amnionless (cubam receptor) on the terminal ileal enterocyte; (5) absorbed, bound to transcobalamin II in blood | Needs INTACT stomach (IF), functional PANCREAS (R-protein degradation), and TERMINAL ILEUM. Failure at any → B₁₂ deficiency (megaloblastic anaemia, subacute combined degeneration of the cord). Causes: pernicious anaemia (anti-parietal/IF), gastrectomy, pancreatic insufficiency, terminal ileal disease/resection |
| Iron (Fe²⁺ ferrous form) | Duodenum/proximal jejunum | Gastric acid reduces Fe³⁺→Fe²⁺ and solubilises; DMT1 (divalent metal transporter) on the apical membrane; basolateral ferroportin exports Fe²⁺; hepcidin (liver, in inflammation) degrades ferroportin → iron trapping in enterocytes/macrophages → anaemia of chronic disease | Needs acid (PPIs/atrophic gastritis impair) and reducing conditions. Hepcidin is the master iron regulator; inflammation-driven hepcidin excess explains why ICU anaemia of chronic disease has HIGH ferritin (trapped iron) and LOW iron saturation [7] |
| Calcium | Duodenum (active), jejunum/ileum (passive) | Active vitamin D-dependent transcellular transport: 1,25-(OH)₂-D (calcitriol) induces the apical TRPV6 channel → calbindin-D9k shuttles Ca²⁺ → basolateral Ca²⁺-ATPase (PMCA) / NCX. Passive paracellular when intake high | Vitamin D-dependent; cholestasis (fat-soluble vitamin malabsorption) → hypocalcaemia, osteomalacia. Needs the acidic duodenal milieu |
| Folate | Jejunum | Reduced and absorbed by reduced folate carrier | Malabsorbed in coeliac/tropical sprue → megaloblastic anaemia (distinct from B₁₂: no neuropathy) |
| Bile acids | Terminal ileum (active, ASBT) | The enterohepatic circulation | Terminal ileal resection → bile acid diarrhoea, steatorrhoea, gallstones |
The two classic terminal-ileum dependencies — vitamin B₁₂ and bile acids — are the most examined absorption facts: any disease or resection of the terminal ileum causes B₁₂ deficiency (megaloblastic anaemia ± neuropathy) and bile acid malabsorption (diarrhoea, steatorrhoea, gallstones).[7][4]
Gut motility — the migrating motor complex and peristalsis
Gut motility serves two distinct modes — the fed pattern (mixing and slow propulsive movement to allow absorption) and the fasting pattern (housekeeping to clear residual content and bacteria). The switch between them is hormonally controlled (motilin initiates fasting activity). [1]
Fed vs fasting (interdigestive) motility
| Feature | Fed pattern | Fasting pattern — the MIGRATING MOTOR COMPLEX (MMC) |
|---|---|---|
| Trigger | Ingestion of a meal (gastrin, CCK, insulin) | Motilin (M cells of duodenum, cycling every ~90 min in the fasted state) |
| Pattern | Segmentation (mixing) + low-amplitude peristalsis | Organised, propagating peristaltic wave sweeping from stomach → through small bowel → to terminal ileum |
| Function | Mix chyme with enzymes, slow propulsion to maximise absorption | "Housekeeper" wave — clears residual food, mucus, desquamated cells and bacteria from the upper gut, preventing bacterial overgrowth |
| Clinical relevance | Fed state SUPPRESSES the MMC | Loss of the MMC (critical illness, opioids, diabetes, post-operative ileus) → stasis → small intestinal bacterial overgrowth (SIBO). Erythromycin (motilin agonist) prokinetic |
The MMC has three phases that recur every ~90-120 minutes in the fasted state: Phase I (quiescence), Phase II (irregular contractions of increasing amplitude), Phase III (the intense, short burst of regular, high-amplitude peristaltic contractions — the "housekeeper" wave that does the clearing), and Phase IV (declining activity back to Phase I). The cycle is driven by motilin and the interstitial cells of Cajal (the gut pacemakers, generating slow waves). The MMC is abolished by feeding and by opioids, and is restored by motilin agonists (erythromycin) and the acetylcholinesterase inhibitor neostigmine.[3]
Peristalsis — the coordinated reflex contraction of circular smooth muscle behind a bolus and relaxation ahead of it — is mediated by the enteric nervous system (the intrinsic primary afferent neurons, interneurons, and motor neurons of Auerbach's/myenteric plexus). It functions even after extrinsic denervation (the gut "brain"). The law of the intestine (Bayliss and Starling): distension evokes contraction proximal (oral) and relaxation distal (anal) to the bolus, propelling it aborally. The gastrocolic reflex (mass colonic movement after a meal, mediated by gastrin/CCK and vagal reflexes) is the physiological basis for post-prandial defaecation and for the post-prandial urgency of irritable bowel syndrome.[1]
Splanchnic circulation — the dual supply and the "canary" organ
The splanchnic bed receives ~25% of cardiac output and is the only circulation with two capillary beds in series joined by a portal vein — gut capillaries → portal vein → hepatic sinusoids. The liver's dual blood supply is the central exam fact:[13]
The liver's dual blood supply
| Feature | Portal vein (~75% of flow) | Hepatic artery (~25% of flow) |
|---|---|---|
| Origin | Confluence of superior mesenteric + splenic veins (drains gut, spleen, pancreas, gallbladder) | Branch of the coeliac trunk (common hepatic → proper hepatic) |
| Oxygen content | Partially deoxygenated (nutrient-rich from the gut) | Fully oxygenated |
| Oxygen contribution | ~50-60% of hepatic O₂ supply (large flow despite lower saturation) | ~40-50% of hepatic O₂ supply (high saturation) |
| Pressure | Low (~5-10 mmHg) | Systemic arterial |
| Regulation | Passive — follows gut blood flow | Autoregulated; the hepatic arterial buffer response (HABR) — if portal flow falls, adenosine accumulates and hepatic arterial flow RISES to buffer total hepatic flow |
| Clinical | Portal hypertension (cirrhosis) → ascites, varices, splenomegaly; portal vein thrombosis | Hepatic artery thrombosis (post-transplant) → graft loss |
The gut is a sacrificial circulation: in any shock state, sympathetic (α₁), angiotensin II, vasopressin (V₁) and endothelin preferentially constrict the splanchnic arterioles to divert cardiac output to heart and brain. The gut therefore becomes ischaemic EARLY and remains so LONG after systemic perfusion is restored (the splanchnic debt repaid last) — the physiological basis for mesenteric ischaemia complicating any shock, and for the persistent lactate that signals ongoing splanchnic hypoperfusion despite a "normal" MAP. The colonic watershed areas (splenic flexure / Griffith's point at the SMA-IMA junction; rectosigmoid / Sudeck's point at the IMA-internal iliac junction) have the poorest collaterals and are most vulnerable to ischaemic colitis.[13]
The gut barrier — the gatekeeper that fails in critical illness
The gut barrier (~300 m² surface area) must simultaneously absorb nutrients and exclude ~10¹³ luminal organisms. It has four integrated layers: (1) the mucus layer (goblet-cell mucin-2 + secretory IgA); (2) the epithelium + tight junctions (claudins, occludin, ZO-1; Paneth-cell antimicrobial peptides — defensins, RegIIIγ; turnover every 3-5 days, the fastest-renewing epithelium); (3) GALT (Peyer's patches, lamina propria lymphocytes, intraepithelial lymphocytes — ~70% of body immune cells); and (4) the microbiome (~10¹³ organisms, dominated by anaerobes, providing colonisation resistance). The barrier is disrupted in critical illness by ischaemia-reperfusion, antibiotics (dysbiosis), PPIs (bacterial overgrowth), ileus, and parenteral nutrition → bacterial/endotoxin translocation → Kupffer-cell cytokine cascade → MODS — the "gut motor" of MODS. The cycle is broken by restoring perfusion early, early enteral nutrition (maintains mucosal integrity and trophic flow), and antibiotic/PPI stewardship.[12][14]
Hepatic physiology
- The bilirubin metabolism — the heme (the - the - the) → the biliverdin → the unconjugated the bilirubin (the lipid-soluble; the albumin-bound) → the liver → the conjugation (the glucuronic acid; the water-soluble) → the bile → the gut → the urobilinogen → the stercobilin (the stool) and the - the - the.[1]
- The urea cycle — the ammonia (the toxic) → the urea (the - the - the - the).[1]
- The glucose homeostasis — the glycogen the storage; the gluconeogenesis; the glycogenolysis.[1]
- The coagulation — the factors II, VII, IX, X (the vitamin K dependent); the factor V; the albumin.[1]
- The detoxification — the phase I (the cytochrome P450) and the phase II (the conjugation).[1]
- The bile — the bile salts (the emulsify); the cholesterol; the bilirubin.[1]
The liver as a metabolic factory — overview
The liver weighs ~1.5 kg and performs >500 functions. For the intensivist, six are central: the dual blood supply, glucose homeostasis, protein synthesis, lipid metabolism, drug metabolism, bilirubin metabolism, the urea cycle / ammonia detoxification, and regeneration. Each is examined in detail below.[1]
Glucose homeostasis — the liver as glycostat
The liver maintains plasma glucose in the narrow 4-7 mmol/L range by balancing storage, mobilisation and new synthesis. It stores ~100 g glycogen (enough for ~12-24 h of fasting) and is the only organ (with the kidney) that can release free glucose into the blood (it expresses glucose-6-phosphatase; muscle does not, so muscle glycogen feeds only itself via lactate).[1]
Hepatic glucose handling — fed vs fasted / stressed state
| Process | Fed state (insulin dominant) | Fasted / stressed (glucagon, catecholamines, cortisol dominant) |
|---|---|---|
| Glycogenesis | Glucose → glycogen (glycogen synthase active, stimulated by insulin) | Suppressed |
| Glycogenolysis | Suppressed | Glycogen → glucose-1-P → glucose (glycogen phosphorylase active) — the FIRST-line glucose source, exhausts stores in ~12-24 h |
| Gluconeogenesis | Suppressed | NEW glucose from lactate (Cori cycle), glycerol (lipolysis), amino acids (alanine → pyruvate via ALT) — the MAIN source in prolonged fasting and critical illness; driven by glucagon (↑ PEPCK, fructose-1,6-bisphosphatase, glucose-6-phosphatase) |
| Net effect | Glucose UPTAKE and storage | Glucose OUTPUT to feed the brain (obligate glucose consumer, ~120 g/day) |
In critical illness the liver is pushed to maximal gluconeogenesis by the counter-regulatory surge (cortisol, catecholamines, glucagon, GH overriding insulin) — the stress hyperglycaemia of sepsis/trauma/burns, fuelled by substrate (lactate from hypoperfused tissues, amino acids from muscle catabolism, glycerol from lipolysis). Two failure modes matter in ICU: (a) hypoglycaemia in acute liver failure (exhausted glycogen + failed gluconeogenesis + impaired insulin clearance → hyperinsulinaemia) — check glucose hourly, it is masked by encephalopathy; (b) refeeding syndrome — when nutrition restarts after starvation, the insulin surge drives phosphate, magnesium and potassium intracellularly for glycolysis/ATP phosphorylation → dangerous hypophosphataemia.[1]
Protein synthesis — the liver makes most plasma proteins
The liver synthesises the majority of circulating proteins. The clotting factors are the most exam-relevant because their synthesis is the basis for monitoring liver synthetic function: [1]
Hepatic protein synthesis — the clinically critical products
| Protein | Function | Half-life | Clinical relevance in liver failure |
|---|---|---|---|
| Albumin | Oncotic pressure (75% of colloid oncotic), drug/bilirubin binding, antioxidant, endothelial stabiliser | ~20 days | Low albumin → oedema, ascites, altered drug binding. LONG half-life → albumin is a LATE marker of synthetic failure |
| Clotting factors II, VII, IX, X | Coagulation (II, VII, IX, X are vitamin K-dependent, needing γ-carboxylation) | Factor VII ~6 h (SHORTEST) | The EARLIEST marker of synthetic failure — factor VII has the shortest half-life → PT/INR rises FIRST (the basis of INR in King's College Criteria). Corrects with vitamin K if cholestatic, NOT if hepatocellular (no functioning hepatocytes to carboxylate) |
| Factor V | Common-pathway cofactor (NOT vitamin-K-dependent) | ~12-15 h | Falls in pure hepatocellular failure; factor V distinguishes hepatocellular (↓) from cholestatic/vitamin-K-deficiency (preserved) coagulopathy |
| Fibrinogen (factor I) | Acute-phase reactant; final coagulation substrate | ~4 days | Often NORMAL/RAISED in acute illness (acute-phase response); a LOW fibrinogen in liver disease implies decompensation or DIC |
| Factor VIII, vWF | Intrinsic pathway / platelet adhesion | — | Paradoxically NORMAL/RAISED in liver failure (made by endothelium, not hepatocytes) → the "rebalanced haemostasis" of cirrhosis (risk of portal vein thrombosis despite high INR) |
| Complement (C3, C4) | Innate immunity, opsonisation | — | Reduced in cirrhosis → impaired opsonisation → susceptibility to infection (esp. spontaneous bacterial peritonitis) |
The synthesis of ALL clotting factors EXCEPT factor VIII and von Willebrand factor (both endothelial) is hepatic. The synthesis sequence that fails in liver disease is clinically useful: clotting (factor VII, half-life 6 h) first, then ammonia clearance (encephalopathy), then albumin (half-life 20 days) last — a graded window onto the severity and chronicity of hepatic synthetic failure.[1]
Lipid metabolism — β-oxidation, ketogenesis, lipoprotein synthesis
The liver is the hub of lipid metabolism: it (1) β-oxidises free fatty acids (released from adipose tissue by glucagon-sensitive hormone-sensitive lipase in the fasted state) → acetyl-CoA → ATP (energy) and ketone bodies (acetoacetate, β-hydroxybutyrate) — ketogenesis, the brain's alternative fuel in prolonged fasting; (2) synthesises fatty acids and triglycerides in the fed state; (3) synthesises and secretes the apolipoproteins and packages VLDL (endogenous triglyceride transport), HDL, and accepts chylomicron remnants; and (4) synthesises cholesterol and converts it to bile acids (the major cholesterol disposal route).[1]
The fasted/stressed ICU patient runs on hepatic β-oxidation and ketogenesis: lipolysis (driven by counter-regulatory hormones) floods the liver with free fatty acids → β-oxidation → acetyl-CoA → ketones exported to brain, heart and muscle. In fatty liver of critical illness / sepsis (and in alcoholic and non-alcoholic fatty liver disease), triglyceride accumulation outstrips β-oxidation/export → steatosis → mitochondrial dysfunction → progression to steatohepatitis and cirrhosis.[1]
Drug metabolism — Phase I (CYP450) and Phase II (conjugation)
The liver converts lipophilic drugs (which would accumulate in fat and resist renal excretion) into polar, water-soluble metabolites for elimination. This occurs in two phases: [1]
Hepatic drug metabolism — Phase I vs Phase II
| Feature | Phase I | Phase II |
|---|---|---|
| Reaction | Oxidation, reduction, hydrolysis — adds/exposes a functional group (-OH, -NH₂, -SH, -COOH) | Conjugation — attaches a polar endogenous moiety to the functional group |
| Enzyme system | Cytochrome P450 superfamily (haem-thiolate, smooth ER) — CYP3A4 (~50% of drugs), CYP2D6, CYP1A2, CYP2C9, CYP2E1 | Transferases — UGT (glucuronidation), sulfotransferases, N-acetyltransferases, glutathione-S-transferases |
| Effect on drug | Often ACTIVATES (prodrug → active: codeine → morphine via CYP2D6) or generates REACTIVE/toxic intermediates (paracetamol → NAPQI via CYP2E1) | Almost always INACTIVATES and increases water-solubility for renal/biliary excretion |
| Effect of cirrhosis | MARKEDLY impaired (↓ CYP content + portosystemic shunting abolishes first-pass) → drug accumulation | Relatively PRESERVED early (glucuronidation less affected) but eventually impaired — the basis for preferring lorazepam (glucuronidated) over diazepam (oxidised) in hepatic failure |
| Inducers | Phenobarbitone, rifampicin, carbamazepine, phenytoin, alcohol, St John's wort → ↑ metabolism → therapeutic failure | Less commonly induced |
| Inhibitors | Macrolides (erythro/clarithromycin), azole antifungals, grapefruit juice, amiodarone, cimetidine → ↓ metabolism → toxicity | Competitive substrates |
First-pass (presystemic) metabolism: orally absorbed drugs pass via the portal vein through the liver BEFORE reaching the systemic circulation — a fraction is metabolised before it reaches its target (bioavailability < 100%). In cirrhosis, portosystemic shunts bypass the liver → first-pass metabolism is LOST → bioavailability of high-extraction drugs (morphine, midazolam, propranolol, verapamil) rises dramatically → a normal oral dose causes overdose. The rule in liver failure: BOTH phases are impaired but Phase I falls earlier and faster than Phase II — prefer drugs cleared by conjugation, start LOW, go SLOW.[1]
Paracetamol hepatotoxicity is the classic Phase I mechanism: paracetamol is mostly glucuronidated/sulphated (Phase II), but a small fraction undergoes CYP2E1 oxidation → NAPQI, a reactive intermediate normally detoxified by glutathione. In overdose (saturating Phase II) or with chronic alcohol (CYP2E1 induction + glutathione depletion), NAPQI accumulates → centrilobular (zone 3) hepatocellular necrosis → acute liver failure. The antidote N-acetylcysteine (NAC) restores glutathione. The King's College Criteria for paracetamol-induced ALF use pH, INR, creatinine and grade of encephalopathy.[1]
Bilirubin metabolism — unconjugated → conjugated → enterohepatic
Bilirubin is the yellow-orange end-product of haem catabolism. Its metabolism is a classic exam cascade, and derangement at each step produces a recognisable clinical-biochemical pattern. [1]
Bilirubin metabolism — step by step
- HAEM CATABOLISM (reticuloendothelial system — spleen, liver Kupffer cells, bone marrow): senescent RBCs are phagocytosed; haem oxygenase cleaves the porphyrin ring of haem → iron (recycled), carbon monoxide (exhaled — a marker of haem turnover) and biliverdin (green). ~250-300 mg bilirubin produced daily; ~80% from senescent RBCs, ~20% from non-RBC haem proteins and ineffective erythropoiesis.
- BILIVERDIN → UNCONJUGATED BILIRUBIN: biliverdin reductase reduces biliverdin to unconjugated bilirubin (UCB). UCB is FAT-SOLUBLE (non-polar — internal hydrogen bonding hides polar groups), so it travels in plasma bound to albumin and CANNOT cross the glomerulus → NOT excreted in urine (hence "indirect" bilirubin — needs alcohol to dissociate for the van den Bergh reaction). UCB DOES cross the blood-brain barrier and the immature neonatal barrier → kernicterus.
- HEPATOCYTE UPTAKE: UCB dissociates from albumin, taken up at the sinusoidal membrane by OATP transporters, binds intracellular ligandin (Y protein / GSTA) and Z protein for cytosolic solubilisation.
- CONJUGATION (the key hepatic step): in the smooth ER, UDP-glucuronosyltransferase (UGT1A1) conjugates UCB with two molecules of glucuronic acid → bilirubin diglucuronide (conjugated bilirubin, CB). CB is WATER-SOLUBLE (polar) → freely filtered at the glomerulus → excreted in urine (hence "direct" bilirubin — reacts directly in the van den Bergh test). This step FAILS in Gilbert and Crigler-Najjar (UGT1A1 deficiency → unconjugated jaundice) and is overwhelmed in massive haemolysis.
- BILIARY EXCRETION: CB is actively secreted across the canalicular membrane by MRP2 (ABCC2) — the rate-limiting, energy-requiring step, the site of failure in Dubin-Johnson syndrome. CB gives bile its yellow-green colour.
- INTESTINAL FATE — UROBILINOGEN and STERCOBILIN: colonic BACTERIA deconjugate CB back to UCB and reduce it to urobilinogen (colourless). Most urobilinogen is further reduced to stercobilin (brown) and excreted in faeces (giving stool its normal brown colour). ~10% of urobilinogen is reabsorbed — the enterohepatic circulation — returned to the liver via the portal vein → re-excreted in bile OR escapes hepatic uptake and is excreted in urine as urobilin (yellow).
Patterns of jaundice — using the bilirubin cascade
| Pattern | Site of lesion | Serum bilirubin | Urine | Stool | Cause |
|---|---|---|---|---|---|
| Pre-hepatic (haemolytic) | Haem catabolism overwhelmed | ↑ UNCONJUGATED | Normal (no CB) but ↑ urobilinogen | Dark (↑ stercobilin) | Haemolysis, reabsorbed haematoma, ineffective erythropoiesis |
| Hepatic (hepatocellular) | Uptake/conjugation/excretion impaired | ↑ MIXED | Dark (CB present); ↑ urobilinogen | Variable (pale if cholestatic) | Hepatitis, cirrhosis, sepsis, drugs, Gilbert/Crigler-Najjar (isolated UCB) |
| Post-hepatic (obstructive) | Excretion into bile blocked | ↑ CONJUGATED | Dark — bilirubinuria (often the FIRST sign) | Pale / clay-coloured (acholic) — no stercobilin; pruritus | Gallstones, pancreatic cancer, cholangiocarcinoma, PSC; intrahepatic cholestasis (sepsis, drugs) |
The two exam pearls: (1) bilirubinuria (dark urine with conjugated hyperbilirubinaemia) precedes clinical jaundice — CB is water-soluble and filtered before skin threshold is reached; (2) pale stool + dark urine = obstructive cholestasis (conjugated bilirubin cannot reach the gut → no stercobilin → pale stool — and backs up into blood → spills into urine → dark urine).[1]
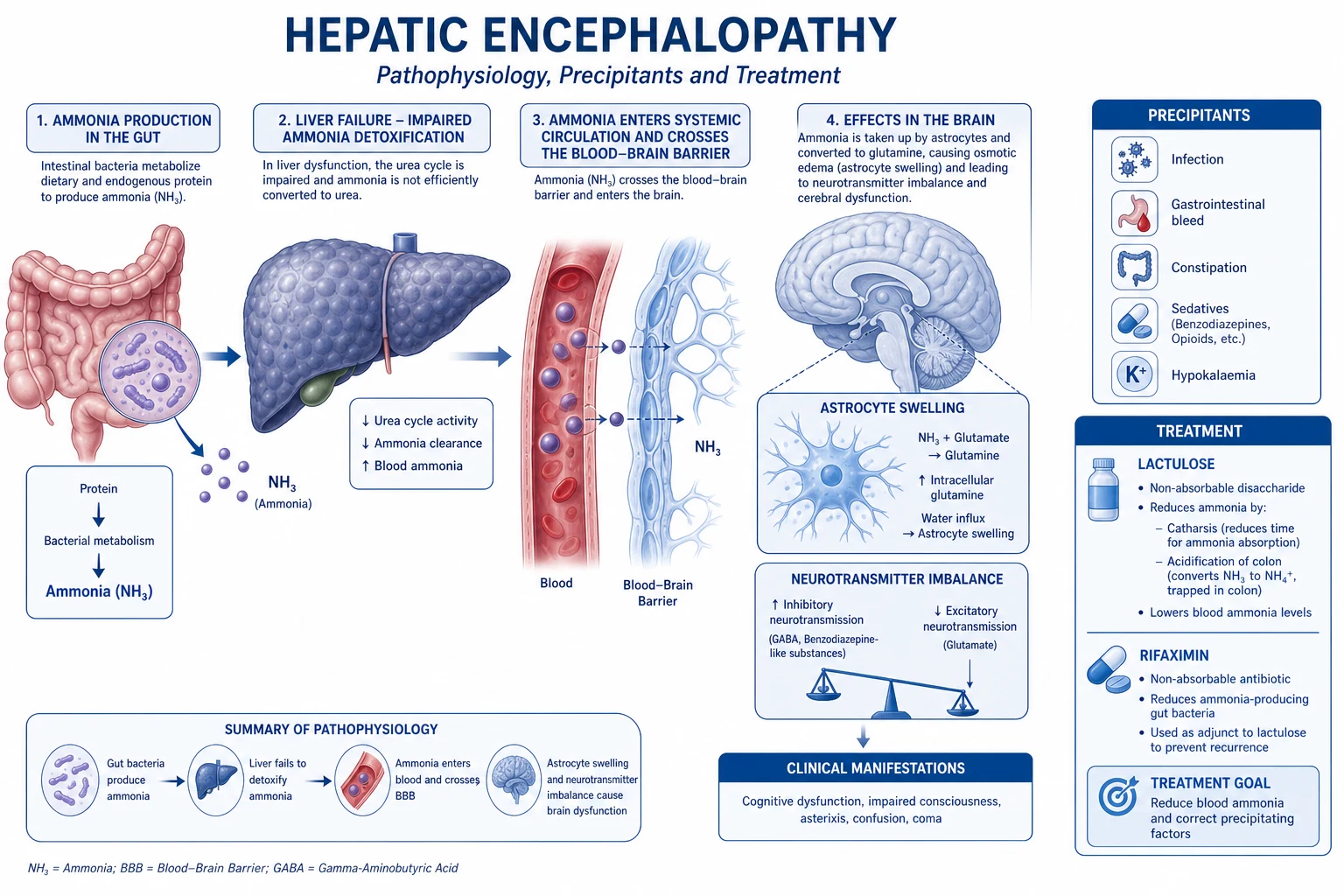
Ammonia → urea cycle — hepatic detoxification
Ammonia (NH₃) is a neurotoxic by-product of nitrogen metabolism. The gut generates most of it (bacterial deamination of dietary protein, urease splitting of urea), and the liver detoxifies it via the urea cycle (Krebs-Henseleit cycle) — the reason portal blood ammonia is high but systemic ammonia is low.[1]
The urea cycle — ammonia to urea
- AMMONIA PRODUCTION — chiefly in the GUT (bacterial urease splits urea → NH₃ + CO₂; bacterial deamination of amino acids → NH₃); the kidney (proximal tubule, glutamine via glutaminase) and muscle (transamination) are minor sources; in catabolism/critical illness muscle becomes a NET producer.
- PORTAL DELIVERY — gut-derived NH₃ enters the portal vein (portal [NH₃] ≫ systemic [NH₃]).
- HEPATIC UREA CYCLE (mitochondria + cytosol): NH₃ + CO₂ + 2 ATP → carbamoyl phosphate (via carbamoyl phosphate synthetase-I, CPS-I — the rate-limiting enzyme, requires N-acetylglutamate as an obligate activator) → + ornithine → citrulline (mitochondria) → + aspartate → argininosuccinate → arginine → arginine + H₂O (arginase) → UREA + ornithine (regenerated). The cycle converts 2 NH₃ + CO₂ → urea (2 nitrogen atoms per urea).
- UREA EXCRETION — urea is water-soluble, leaves the liver in systemic blood → filtered by the kidney → excreted in urine (BUN / serum urea). A fraction is recycled to the gut (split by bacterial urease) — the urea salvage cycle.
- FAILURE → HYPERAMMONAEMIA — when the liver fails (acute or chronic) or portal blood is shunted past the liver (portosystemic shunts, TIPSS), NH₃ accumulates in systemic blood.
- NEUROTOXICITY → HEPATIC ENCEPHALOPATHY — NH₃ crosses the blood-brain barrier → taken up by ASTROCYTES (the only brain cell expressing glutamine synthetase) → NH₃ + glutamate → glutamine → osmotically active → astrocyte swelling → cerebral oedema (in acute liver failure, potentially fatal intracranial hypertension/herniation) and astrocyte dysfunction (Alzheimer type II change) → impaired neurotransmission → encephalopathy.[11]
Liver regeneration — the unique capacity for compensatory hyperplasia
The liver is the only human solid organ with a clinically meaningful regenerative capacity — following injury or resection, the remaining hepatocytes re-enter the cell cycle and restore mass (compensatory hyperplasia, not true regeneration of lobular architecture). Up to 70% of the liver can be resected with full functional recovery in weeks. This is the basis of live-donor liver transplantation (a partial graft regenerates in both donor and recipient) and the recovery potential of acute hepatic injury.[9]
Drivers of liver regeneration
| Phase / signal | Mediators | Notes |
|---|---|---|
| Priming (G₀ → G₁) | TNF-α, IL-6 (from Kupffer cells), via NF-κB and STAT3 | The injury/inflammation signal that "wakes" hepatocytes; HGF is released from stores |
| Progression (cell-cycle progression) | HGF (hepatocyte growth factor), EGF (epidermal growth factor), TGF-α | The mitogens driving DNA synthesis and mitosis |
| Termination | TGF-β (the most potent inhibitor), activin | Stops proliferation once mass is restored |
| Auxiliary cells | Hepatic stellate cells, endothelial cells, oval/progenitor cells | Provide HGF, scaffold the rebuilt vasculature, contribute when mature hepatocytes cannot (chronic injury) |
Regeneration FAILS in chronic injury (cirrhosis, fibrosis — the stellate-cell collagen scar distorts the architecture and blocks regeneration), in senescence, and in massive acute injury beyond the replicative reserve → acute liver failure. The regenerative drive also explains hepatocellular carcinoma (chronic injury → chronic regenerative cycling → mutational accumulation → carcinoma in cirrhotic livers).[9]
Hepatic encephalopathy mechanism — the ammonia-astrocyte-oedema axis
Hepatic encephalopathy (HE) is the reversible spectrum of neuropsychiatric disturbance in liver failure, ranging from inattention/mild confusion through asterixis (flapping tremor) and somnolence to coma. Ammonia is the central, most actionable toxin, but NOT the sole mechanism.[10][11]
The core mechanism: hyperammonaemia → NH₃ crosses the BBB → astrocyte glutamine synthetase → glutamine → osmotic astrocyte swelling → cerebral oedema + astrocyte dysfunction. In ACUTE liver failure this is the leading cause of death (intracranial hypertension, uncal herniation) — an arterial ammonia > 150 µmol/L with high-grade encephalopathy predicts cerebral oedema/herniation. In CIRRHOSIS (chronic, with portosystemic shunting), chronic low-grade ammonia exposure produces the Alzheimer type II astrocyte change and a predominantly GABA-ergic (increased inhibitory) neurotransmission picture (hence the historical response to flumazenil).[11]
Additional contributors beyond ammonia: systemic inflammation (cytokines increase BBB permeability and ammonia sensitivity), benzodiazepine-like compounds (from the gut), manganese (basal ganglia deposition in chronic portosystemic shunting), and mercaptans. The clinical management follows the physiology: lactulose (acidifies the colon → traps NH₃ as non-absorbable NH₄⁺ → laxation expels it) and rifaximin (a minimally-absorbed antibiotic that suppresses urease-producing bacteria) — ammonia-lowering is the backbone of therapy. Precipitants of HE in cirrhosis (GI bleed → protein load; infection; constipation; electrolyte disturbance; sedatives; TIPSS → increased portosystemic shunting) all share the mechanism of acutely increasing ammonia or BBB permeability.[10]
Clinical correlations
- The jaundice (the pre-hepatic — the haemolysis; the hepatic — the hepatocellular; the post-hepatic — the obstruction).[1]
- The hepatic encephalopathy (the ammonia).[1]
- The coagulopathy (the factors).[1]
- The portal hypertension (the cirrhosis; the varices; the ascites).[1]
Exam practice — SAQs
SAQ — Splanchnic circulation in shock: a persistent lactate and the ischaemic gut
10 minutes · 10 marks
A 68-year-old man is admitted to ICU 12 hours after an emergency Hartmann procedure for a perforated sigmoid diverticulum. He remains in septic shock despite 30 mL/kg crystalloid: MAP 62 on noradrenaline 0.35 mcg/kg/min, lactate risen from 3.2 to 5.8 mmol/L, urine output 10 mL/h. He is intubated and ventilated (FiO2 0.6) with cool peripheries, a distended silent abdomen and coffee-ground nasogastric aspirate. AST 480, ALT 410 (baseline normal), INR 2.1, bilirubin 52. The registrar asks why the lactate keeps climbing when the blood pressure is acceptable.
Clinical pearls
Red flags
Prognosis and integrated physiology
Key physiological determinants and clinical outcomes
| Scenario | Mechanism / outcome | Notes |
|---|---|---|
| Mesenteric ischaemia (any shock) | Mortality 50-80% if transmural infarction; gut is the 'canary' of the splanchnic bed | Restore perfusion EARLY; lactate is the sentinel marker of ongoing splanchnic hypoperfusion |
| Gut barrier disruption → translocation | Drives MODS mortality; the 'second hit' — a subsequent insult in a primed host produces exaggerated inflammation | Prevent: restore perfusion, early enteral nutrition, antibiotic/PPI stewardship |
| Acute liver failure — cerebral oedema | Leading cause of death; arterial ammonia >150 + grade III-IV encephalopathy = high risk | King's College criteria trigger transplant referral; mortality ~30-50% without transplant |
| Cirrhosis — drug handling | Accumulation of sedatives/opioids → oversedation, hypotension, encephalopathy | Both Phase I and II impaired; first-pass lost; start LOW, go SLOW |
| Cholestasis | Reversible if cause removed (sepsis, drugs, obstruction relieved) | Fat-soluble vitamin malabsorption → coagulopathy (vit K), bone disease (vit D); pruritus often the most distressing symptom |
| Acute pancreatitis (zymogen activation) | Premature intracellular trypsinogen activation → autodigestion | The physiological safeguard (PSTI/SPINK1 trypsin inhibitor, zymogen packaging) fails |
| Major hepatectomy (to 70%) | Full functional recovery in weeks via regeneration | TNF-α/IL-6 prime, HGF/EGF progress, TGF-β terminates; fails in cirrhosis |
The unifying principle is that the gut-liver axis is BOTH a target of, and a driver of, critical illness. In shock the sacrificed splanchnic circulation becomes ischaemic, the gut barrier fails, and bacterial/endotoxin translocation activates the Kupffer-cell cytokine cascade that propagates inflammation to distant organs — the 'gut motor' of MODS. The failing liver loses its synthetic (clotting factors, albumin), metabolic (glucose, ammonia, drug clearance), and excretory (bilirubin, bile) functions in a predictable sequence — clotting (factor VII, half-life 6 h) first, then ammonia/encephalopathy, then albumin (half-life 20 days) last. Understanding this physiology — the dual blood supply and hepatic arterial buffer response, the three phases of gastric acid secretion, the zymogen activation safeguard, the enterohepatic circulation, the terminal-ileum dependencies (B₁₂ and bile acids), the migrating motor complex, the phases of drug metabolism, the bilirubin cascade, the urea cycle, liver regeneration, and the ammonia-astrocyte-oedema axis of encephalopathy — is the foundation for every ICU intervention from early enteral feeding and antibiotic stewardship to PPI selection, ammonia-lowering therapy, neuroprotection in liver failure, and cholestasis management.[1][12][11]
Key trials and evidence
Hofmann & Hagey 2014 — Key discoveries in bile acid chemistry and biology (J Lipid Res; PMID 24838141)
Source
Authoritative history and synthesis of eight decades of bile acid biochemistry by Alan Hofmann, the field's principal architect
Key contribution
Definitive account of bile acid synthesis from cholesterol (7α-hydroxylase, CYP7A1), conjugation, and the enterohepatic circulation — the framework for understanding fat digestion, gallstone pathogenesis, and bile acid malabsorption
Key finding
The bile acid pool (~3 g) cycles 6-10 times/day with 95% ileal reabsorption; only ~0.5 g is synthesised daily — the pool is preserved by efficient recycling, and lost when the terminal ileum is diseased/resected
Clinical bottom line
The physiological basis for fat malabsorption, steatorrhoea, choleretic diarrhoea and cholesterol gallstones after terminal ileal disease/resection, and for the bile acid sequestrants in bile-acid diarrhoea
Schubert 2017 — Regulation of gastric acid secretion (Curr Opin Gastroenterol; PMID 28787289)
Source
Contemporary review of the physiologic, pathophysiologic and pharmacologic regulation of gastric acid secretion
Key contribution
Integrated the three secretagogues (histamine-H₂, gastrin-CCKB, ACh-M₃) and the master inhibitor somatostatin at the parietal cell, with the H⁺/K⁺-ATPase (proton pump) as the final common effector
Key finding
The parietal-cell activation cascade (tubulovesicle-to-canalicular translocation of the pump on stimulation) explains why PPIs only inactivate ACTIVATED pumps — the rationale for dosing ~30 min before a meal
Clinical bottom line
The mechanistic basis for H₂ blockers, PPIs and somatostatin analogues, and for hypergastrinaemia and SIBO with chronic acid suppression
Takahashi 2013 — The migrating motor complex (J Smooth Muscle Res; PMID 24662475)
Source
Comprehensive review of the mechanism and clinical importance of the interdigestive migrating motor complex
Key contribution
Defined the three-phase MMC (quiescence → irregular → phase-III 'housekeeper' wave), its motilin-driven ~90-min cycling, and the role of the interstitial cells of Cajal as gut pacemakers
Key finding
Loss of the MMC (critical illness, opioids, diabetes, post-operative ileus) → stasis → small intestinal bacterial overgrowth; motilin agonists (erythromycin) and neostigmine restore it
Clinical bottom line
The physiological rationale for prokinetics in gastroparesis/ileus and for understanding SIBO in the critically ill
Michalopoulos 2014 — Advances in liver regeneration (Expert Rev Gastroenterol Hepatol; PMID 24964729)
Source
Review of the molecular mechanisms of liver regeneration by the leading investigator in the field
Key contribution
Defined the priming (TNF-α/IL-6 → NF-κB/STAT3) and progression (HGF, EGF, TGF-α) signals and the termination signal (TGF-β), with the hepatic stellate and oval/progenitor cell contributions
Key finding
Up to 70% of the liver can be resected with full functional recovery in weeks — the basis of live-donor liver transplantation; regeneration fails in cirrhosis (scar distorts architecture)
Clinical bottom line
The recovery potential of acute hepatic injury and the regenerative basis of hepatocellular carcinoma in chronically-injured cirrhotic livers
Deitch 2012 — Gut-origin sepsis: evolution of a concept (Surgeon; PMID 22534256)
Source
Seminal review by Edwin Deitch, the leading proponent of the gut-origin theory, synthesising four decades of work
Key contribution
Articulated the 'gut as the motor of MODS' — that the ischaemic, barrier-disrupted gut in shock is not merely a victim but a DRIVER of distant organ failure via bacterial/endotoxin translocation
Key finding
Two routes carry gut-derived noxious material systemically: the PORTAL route (→ Kupffer cells → cytokines) and the MESENTERIC LYMPH route (→ thoracic duct → systemic, bypassing the liver)
Clinical bottom line
The physiological rationale for protecting the gut in critical illness — early perfusion, early enteral nutrition, antibiotic/PPI stewardship — as a MODS-prevention strategy
References
- [1]Schubert ML Physiologic, pathophysiologic, and pharmacologic regulation of gastric acid secretion Curr Opin Gastroenterol, 2017.PMID 28787289
- [2]Schubert ML, Rehfeld JF Gastric Peptides-Gastrin and Somatostatin Compr Physiol, 2019.PMID 31853955
- [3]Takahashi T Interdigestive migrating motor complex -its mechanism and clinical importance J Smooth Muscle Res, 2013.PMID 24662475
- [4]Hofmann AF, Hagey LR Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades J Lipid Res, 2014.PMID 24838141
- [5]Hofmann AF The enterohepatic circulation of bile acids in mammals: form and functions Front Biosci (Landmark Ed), 2009.PMID 19273221
- [6]Hegyi P, Maleth J, Walters JR Guts and Gall: Bile Acids in Regulation of Intestinal Epithelial Function in Health and Disease Physiol Rev, 2018.PMID 30067158
- [7]Miret S, Simpson RJ, McKie AT Physiology and molecular biology of dietary iron absorption Annu Rev Nutr, 2003.PMID 12626689
- [8]Weiss FU, Halangk W, Lerch MM New advances in pancreatic cell physiology and pathophysiology Best Pract Res Clin Gastroenterol, 2008.PMID 18206809
- [9]Michalopoulos GK Advances in liver regeneration Expert Rev Gastroenterol Hepatol, 2014.PMID 24964729
- [10]Thabut D, Bouzbib C, Meunier L Diagnosis and management of hepatic encephalopathy: The French recommendations Liver Int, 2023.PMID 36625084
- [11]Blei AT Brain edema in acute liver failure Crit Care Clin, 2008.PMID 18241781
- [12]Deitch EA Gut-origin sepsis: evolution of a concept Surgeon, 2012.PMID 22534256
- [13]Landow L, Andersen LW Splanchnic ischaemia and its role in multiple organ failure Acta Anaesthesiol Scand, 1994.PMID 7839769
- [14]Balzan S, de Almeida Quadros C, de Cleva R, Zilberstein B, Cecconello I Bacterial translocation: overview of mechanisms and clinical impact J Gastroenterol Hepatol, 2007.PMID 17376034