ICU · Neurocritical Care
Acute intermittent porphyria (AIP)
Also known as Acute intermittent porphyria (AIP) · Porphyric crisis · Swedish porphyria · Hemin therapy
AIP is a rare autosomal dominant defect in hepatic haem biosynthesis caused by deficient porphobilinogen deaminase (hydroxymethylbilane synthase, HMBS). Presents with acute neurovisceral attacks triggered by drugs (barbiturates, sulfonamides, hormonal contraceptives, antiepileptics, alcohol), fasting, infection, surgery and stress. Classic triad: abdominal pain (severe, out of proportion to exam, with a benign abdomen) + neurological symptoms (motor neuropathy, seizures, psychiatric disturbance) + autonomic dysfunction (tachycardia, hypertension, constipation, urinary retention). Urine turns dark/red on light exposure. Diagnosis: markedly elevated urinary porphobilinogen (PBG) and aminolevulinic acid (ALA) on a random spot urine, confirmed by HMBS genetic testing. Treatment: withdraw all triggers, IV haem arginate 3 mg/kg once daily for 4 days (or glucose loading ~300 g/day if haem unavailable), opiate analgesia, phenothiazines/ondansetron for nausea, gabapentin/levetiracetam for seizures, and correct SIADH hyponatraemia. Ventilation may be needed for motor neuropathy.
On this page & tools
Your progress
Saved locally on this device.
Target exams
Red flags

Pathophysiology
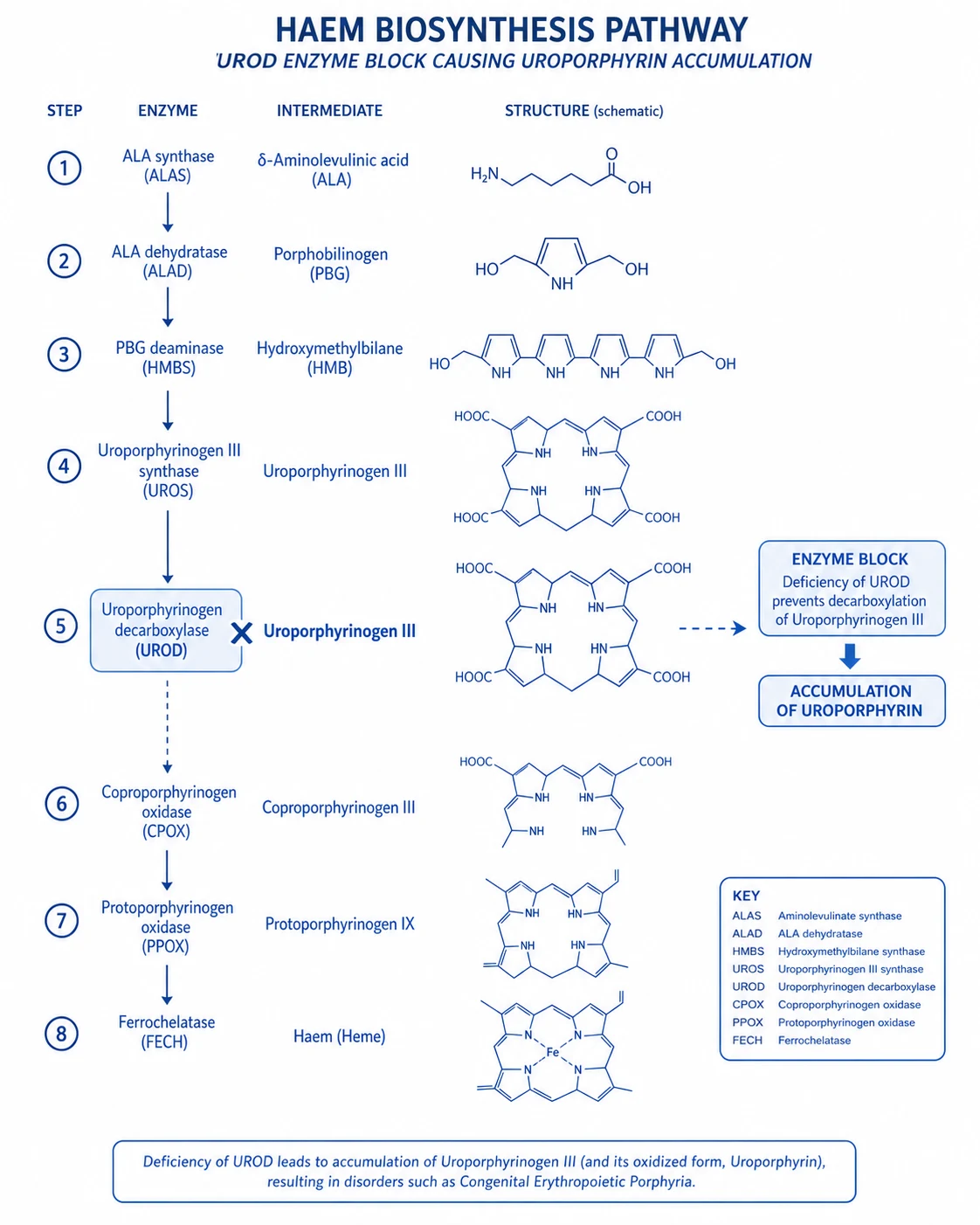
Acute intermittent porphyria (AIP) is the commonest of the acute (neurovisceral) hepatic porphyrias. It arises from a partial (~50%) deficiency of the third enzyme of the haem biosynthetic pathway, porphobilinogen deaminase (PBGD; also called hydroxymethylbilane synthase, HMBS, or uroporphyrinogen I synthase; gene HMBS on chromosome 11q23.3). Inheritance is autosomal dominant with low penetrance — only around 10–20 % of mutation carriers ever experience a clinical attack.[2][3]
The haem biosynthetic pathway (CICM First Part favourite)
Haem is synthesised in every nucleated cell, but the liver (~85 %, for cytochrome P450 / haemoproteins) and erythroid marrow (~15 %, for haemoglobin) are the major sites. The pathway has eight steps, beginning and ending in the mitochondrion: [1]
The eight steps of haem biosynthesis — and where AIP blocks
Glycine + succinyl-CoA → δ-aminolaevulinic acid (ALA)
Catalysed by **ALA synthase (ALAS)** in mitochondria. Two isoforms: ALAS1 (ubiquitous, hepatic — the rate-limiting, feedback-regulated enzyme) and ALAS2 (erythroid). ALAS1 is **induced** by drugs, steroids and fasting and **repressed** by haem (negative feedback). This induction is the engine of every acute attack.
2 × ALA → porphobilinogen (PBG)
Catalysed by **ALA dehydratase** (PBG synthase) in the cytosol. Zinc-dependent. Inhibited by lead (causes a clinically similar neurovisceral syndrome).
4 × PBG → hydroxymethylbilane (HMB)
Catalysed by **porphobilinogen deaminase (HMBS / HMB synthase)** in the cytosol. ⚠️ **THIS IS THE DEFICIENT ENZYME IN AIP.** Activity is reduced to ~50 %, so when ALAS1 is upregulated the pathway backs up and the soluble precursors ALA and PBG accumulate and spill into urine.
HMB → uroporphyrinogen III
Catalysed by uroporphyrinogen III synthase. (Deficiency → congenital erythropoietic porphyria, Günther disease.)
Uroporphyrinogen III → coproporphyrinogen III
Catalysed by uroporphyrinogen decarboxylase. (Deficiency → porphyria cutanea tarda — photosensitivity, no neurovisceral attacks.)
Coproporphyrinogen III → protoporphyrinogen IX
Catalysed by **coproporphyrinogen oxidase** in mitochondria. (Deficiency → **hereditary coproporphyria** — neurovisceral + photosensitivity.)
Protoporphyrinogen IX → protoporphyrin IX
Catalysed by **protoporphyrinogen oxidase**. (Deficiency → **variegate porphyria** — neurovisceral + photosensitivity; common in South Africa.)
Protoporphyrin IX + Fe²⁺ → haem
Catalysed by **ferrochelatase** in mitochondria. (Deficiency → erythropoietic protoporphyria.) Haem then feeds back to repress ALAS1 — the lever that IV haem arginate and glucose both pull.
Why a partial enzyme deficiency only causes intermittent attacks
Because HMBS activity is halved but not abolished, baseline haem production is usually adequate and the carrier is asymptomatic. An attack requires a second hit — an inducer that upregulates hepatic ALAS1. Induction of hepatic cytochrome P450 enzymes (by barbiturates, phenytoin, rifampicin, alcohol, sulphonal compounds) consumes haem; the fall in the free hepatic haem pool de-represses ALAS1. With ALAS1 driven hard against a 50 %-deficient HMBS step, ALA and PBG accumulate massively (urinary PBG rises 5–100×). The same logic explains the three therapeutic levers:[2][4]
- Withdraw the inducer (stops the drive on ALAS1).
- Give IV haem arginate (restores the free haem pool → re-represses ALAS1).
- Give glucose (carbohydrate loading directly represses ALAS1 via the PGC-1α / AMPK axis — "the glucose effect"). [1]
Why the damage is neurological (and why the abdomen is "benign")
AIP produces no skin photosensitivity (unlike variegate, hereditary coproporphyria, PCT) because the accumulated porphyrin precursors are not present in sufficient photoactive quantity in skin. The clinical damage is purely neurovisceral. Several mechanisms are proposed:[5]
- Direct ALA neurotoxicity. ALA is structurally a GABA analogue and is taken up by neural tissue. It perturbs GABAergic neurotransmission, generates reactive oxygen species via iron-mediated Fenton chemistry, and impairs mitochondrial function. PBG itself is less clearly neurotoxic.
- Localised neural haem depletion. Neuronal cells (especially autonomic ganglia and motor neurons) depend on local haem synthesis for respiratory-chain and cytochrome function; ALAS1-driven flux depletes neuronal haem faster than the liver can compensate.
- Autonomic neuropathy of splanchnic viscera. The severe abdominal pain reflects autonomic dysfunction and transient gut dysmotility/vasospasm rather than peritoneal inflammation — explaining the cardinal mismatch of excruciating pain with a soft, non-tender, afebrile abdomen (no guarding, no peritonism).
- Hypothalamic involvement drives inappropriate ADH release → SIADH and hyponatraemia, itself a seizure risk. [1]
Epidemiology and genetics
Frequency
How common
- Mutation prevalence ≈ 1 in 1700–2000 in Northern European populations (founder effects in Sweden, France, South Africa — but the latter is variegate porphyria).
- Clinically manifest AIP is far rarer: 1–5 per 100 000 — penetrance is only ~10–20 %.
- Female : male ratio ≈ 2:1 in symptomatic patients (progesterone/oestrogen drive ALAS1).
- Almost never presents before puberty; peak onset age 20–40 years.
Genetics
Inheritance and testing
- Autosomal dominant; gene = *HMBS* (hydroxymethylbilane synthase) on chromosome 11q23.3.
- >450 disease-causing variants described; most families have private mutations — so genetic counselling and cascade testing of relatives is essential.
- Erythrocyte HMBS enzyme activity is reduced in "classic" AIP but may be normal in rare "variant" forms — **genetic sequencing is the confirmatory test**.
- Screen all first-degree relatives once the proband mutation is known (asymptomatic carriers can avoid triggers).
Triggers — the "5 Ds plus hormones"
Almost every attack can be traced to one or more precipitants. Every suspected AIP patient needs an exhaustive drug, dietary and social history. The mnemonic "Drugs, Diet, Drink, Disease, Debt (stress), plus the Diary (menstrual)" captures them. [1]
DRUGS (the biggest avoidable cause)
Inducers of hepatic ALAS1 / CYP450
- Barbiturates — **THE classic trigger** (thiopentone induction! NEVER in suspected porphyria).
- Antiepileptics — phenytoin, carbamazepine, valproate, phenobarbitone, primidone (avoid all; use gabapentin/levetiracetam).
- Sulfonamides (cotrimoxazole, sulfasalazine).
- Rifampicin, rifabutin.
- Hormonal: combined oral contraceptives, progestogens, anabolic steroids, danazol.
- Ergotamine; metoclopramide (historically listed — use ondansetron instead); glutethimide; griseofulvin; some IMRDs.
- Alcohol — both binge and chronic induction.
Fast / Diet
Carbohydrate depletion derepresses ALAS1
- Crash dieting, rapid weight loss, low-carbohydrate (keto) diets — the "glucose effect" works in reverse: low glucose upregulates ALAS1.
- Prolonged fasting perioperatively or during illness.
- Post-bariatric surgery malabsorption states.
- Treat by avoiding fasting and giving prompt carbohydrate (enteral first, IV dextrose if needed).
Stress / Disease / Surgery
Metabolic demand
- Intercurrent infection (UTI, URTI, pneumonia) — the commonest "non-drug" trigger in hospitalised patients.
- Surgery and general anaesthesia (both fasting + drugs + surgical stress).
- Psychological/physical stress, sleep deprivation, alcohol withdrawal.
- Always plan elective surgery in known AIP with a porphyria-safe anaesthetic plan and avoid pre-op fasting beyond clear fluids.
Menstrual / hormonal
Cyclical attacks
- Luteal-phase (premenstrual) attacks — progesterone potently induces ALAS1.
- Pregnancy is usually well tolerated but postpartum flares occur; contraception is high-risk.
- GnRH analogues (goserelin) with add-back oestrogen can break the menstrual cycle in refractory cyclical disease.
Clinical presentation
The neurovisceral triad
The full-blown attack evolves over hours to days and combines abdominal, neurological and autonomic features, frequently with psychiatric symptoms and hyponatraemia. Abdominal pain is the single most common symptom (>90 % of attacks) and is typically the presenting complaint. [1]
ABDOMINAL (#1 symptom)
Pain out of proportion, benign exam
- Severe, colicky, poorly localised, often diffuse; may radiate to back/loins.
- Out of proportion to a soft, non-tender, afebrile abdomen — NO guarding, NO rebound, NO peritonism.
- Often accompanied by **constipation / ileus**, nausea, vomiting, abdominal distension (autonomic gut dysmotility, not obstruction).
- May mimic a surgical abdomen → negative laparotomy is a well-described misadventure; the scarred abdomen in a young woman is a classic exam clue.
- CT abdomen is typically normal — the value of the scan is to exclude a surgical cause.
Neurological
Predominantly motor
- Motor neuropathy — proximal > distal, can ascend and resemble **Guillain–Barré syndrome**; may progress to tetraparesis and **respiratory failure**.
- Cranial neuropathies — facial (VII), vagal (X), bulbar weakness with aspiration risk; rarely ophthalmoplegia.
- Sensory loss less prominent; patchy, may affect trunk.
- Seizures — both a primary manifestation (up to 20 %) and a consequence of severe hyponatraemia.
- CNS: anxiety, insomnia, depression, hallucinations, paranoia, frank psychosis/delirium (can be the dominant feature).
Autonomic
The "fight or flight" pattern
- **Tachycardia** — the most common sign (~80 %); sinus, often disproportionate to pain/fever.
- Hypertension (sometimes severe), sweating, tremor.
- Constipation / ileus, urinary retention.
- Peripheral vasoconstriction → cool, pale extremities.
SIADH / hyponatraemia
A seizure risk in its own right
- Hypothalamic involvement → inappropriate ADH release; serum Na can fall rapidly to <120 mmol/L.
- Contributes to seizures, coma and agitation; check U&E on every suspected attack.
- Usually resolves as the attack is treated; treat like other SIADH (fluid restriction / hypertonic saline).
The "porphyrin urine" sign
Fresh urine is typically normal in colour but turns dark red, reddish-brown or port-wine on standing/exposure to light, as excess PBG polymerises non-enzymatically to porphobilin and uroporphyrin. A negative dark-urine history does not exclude AIP (depends on concentration and time), but a clearly colour-changing urine is a powerful bedside clue. [1]
Urine that darkens on standing
Suggests porphyria
- Acute intermittent porphyria (AIP) — darkens red/brown.
- Variegate porphyria, hereditary coproporphyria — can also darken.
- Alkaptonuria — darkens black (homogentisic acid).
- PNH / rhabdomyolysis — red/dark from the start (haemoglobin/myoglobin), not light-dependent.
Differential diagnosis
AIP is a renowned "great imitator." The combination of abdominal pain + neurological/psychiatric signs + hyponatraemia should prompt consideration in any atypical or recurrent presentation, especially after a negative surgical workup. [1]
Mimics of the acute abdomen
Exclude surgically
- Perforated viscus / peritonitis — but here the abdomen is hard, with guarding; AIP abdomen is soft.
- Mesenteric ischaemia, pancreatitis, biliary colic, ruptured ectopic.
- Lead colic — also a neurovisceral porphyria-like syndrome (inhibits ALA dehydratase, so ALA ↑ but PBG normal).
- Familial Mediterranean fever — recurrent fevers + serositis, normal PBG.
Mimics of motor neuropathy
Ascending weakness
- Guillain–Barré syndrome — ascending areflexic paralysis; CSF protein↑ (albuminocytologic dissociation). AIP motor neuropathy can be indistinguishable acutely.
- Tick paralysis, botulism, vasculitic neuropathy, critical-illness neuropathy/myopathy.
- Distinguishing clue: AIP has abdominal pain + autonomic storm + elevated urinary PBG.
Psychiatric / seizure mimics
Behavioural change
- Drug/alcohol withdrawal, encephalitis, hypertensive encephalopathy.
- Hyponatraemic delirium from any cause.
- Functional / conversion disorder — a dangerous mislabel: AIP psychosis can land patients in psychiatry before the diagnosis is made.
Diagnosis
First-line during an acute attack: urinary PBG
The single most useful test during an attack is a random spot urine porphobilinogen (PBG) (reported as a PBG:creatinine ratio, so a 24-hour collection is unnecessary). In an acute attack, urinary PBG is markedly elevated (5–100× the upper limit of normal); the result is usually available within 24–48 hours. ALA is also elevated. A normal PBG during an acute symptomatic episode effectively excludes AIP, variegate porphyria and hereditary coproporphyria as the cause.[2][4]
Diagnostic algorithm for a suspected acute attack
1. Random spot urine PBG + ALA (PBG:creatinine ratio)
Send immediately — no 24-h collection needed. Markedly elevated PBG (and ALA) confirms an acute porphyria is in progress. Protect the sample from light if storage is prolonged.
2. Differentiate the acute porphyrias
All three acute porphyrias (AIP, hereditary coproporphyria HCP, variegate porphyria VP) elevate urinary PBG in an attack. To tell them apart send **plasma + faecal porphyrins** and check for **skin photosensitivity**: AIP = no skin lesions; HCP = faecal coproporphyrin ↑; VP = faecal protoporphyrin ↑ and characteristic plasma fluorescence peak at 626 nm. PBG and ALA fall after an attack — so sample early.
3. Confirm with genetic testing
Sequence *HMBS* (and ALAD, CPOX, PPOX if the biochemical pattern is equivocal). Genetic confirmation enables cascade screening of asymptomatic relatives. Do NOT rely on erythrocyte HMBS activity alone — it is normal in rare variant AIP.
4. Between attacks
Urinary PBG may remain elevated for months after an attack, then normalise; a "normal" result in a quiescent carrier does not exclude AIP. Relatives are screened by mutation analysis once the family mutation is known.
5. Supportive / exclusion tests
CT abdomen (exclude surgical abdomen), U&E (SIADH hyponatraemia), LFTs (often mildly deranged; chronic AIP predisposes to cirrhosis and hepatocellular carcinoma), FBC, CK (if weakness), and septic screen to find a trigger. LP if GBS is in the differential.
Biochemical pattern in AIP (during attack)
What the lab shows
- **Urinary PBG ↑↑↑** (5–100× ULN) — the hallmark; random spot sample.
- Urinary ALA ↑↑ (always elevated with PBG in AIP).
- Urinary uroporphyrin ↑ (less specific).
- Faecal porphyrins — usually normal in AIP (this distinguishes AIP from VP and HCP).
- Plasma porphyrins — usually normal in AIP.
Key exclusions / confounders
Don't be fooled
- Lead poisoning — ALA ↑ but **PBG normal** (lead inhibits ALA dehydratase, step 2).
- Hereditary tyrosinaemia type I — ↑ succinylacetone inhibits ALA dehydratase; porphyria-like neurovisceral crises in infants.
- False-positive urinary porphyrins — from any hepatobiliary disease, renal impairment, or urinary tract infection ("porphyrinuria" without porphyria).
- PBG falls after the attack and with haem therapy — sample EARLY.
Management of the acute attack

Treatment must begin on suspicion, before biochemical confirmation, because outcome correlates with speed of haem therapy. The five pillars are: (1) stop triggers, (2) give IV haem arginate, (3) glucose loading if haem is unavailable/delayed, (4) symptom control with porphyria-safe drugs, (5) anticipate and treat complications (hyponatraemia, respiratory failure). [1]
AIP acute attack management — the 5 pillars
1. STOP ALL triggering drugs
Audit the entire medication chart against a current **porphyria-safe drug list** (e.g. the European Porphyria Network / porphyria specialist centre list). Stop: barbiturates, sulfonamides, phenytoin, carbamazepine, valproate, oral contraceptives, rifampicin, alcohol, ergotamine. Substitute with safe alternatives (propofol for sedation, opioids for pain, ondansetron/prochlorperazine for nausea, beta-blockers for hypertension, gabapentin/levetiracetam for seizures).
2. IV haem arginate — specific therapy
**Haem arginate 3 mg/kg IV once daily for 4 days** (US equivalent: hemin/Panhematin 3–4 mg/kg). Reconstitute in human albumin 4–20 % and give via a **central line** to prevent phlebitis/haemolysis. Mechanism: exogenous haem replenishes the hepatic free-haem pool → negative-feedback repression of ALAS1 → falls in ALA and PBG. Give **EARLY** — every day of delay increases attack severity, neuropathy and length of stay. Repeated courses may be needed; watch for coagulopathy (anticoagulant effect of degradation products), phlebitis and iron overload.
3. Glucose loading (if haem unavailable / delayed, or mild attacks)
Carbohydrate inhibits ALAS1 directly (the "glucose effect"). Aim for **~300 g/day** (e.g. 10 % dextrose at 125 mL/h provides ~300 g over 24 h; higher concentrations need a central line). Enteral carbohydrate is preferred when feasible. Monitor: hyperglycaemia, fluid overload, and SIADH hyponatraemia (dextrose is free water — can worsen low Na). Glucose is less effective than haem — use it as a temporising bridge, not a substitute.
4. Symptom control (porphyria-safe drugs only)
**Pain**: opioids are SAFE and usually required in high doses — morphine, fentanyl, oxycodone. **Nausea**: ondansetron, prochlorperazine/phenothiazines. **Seizures**: gabapentin, levetiracetam, magnesium, benzodiazepines — AVOID phenytoin/carbamazepine/valproate/barbiturates. **Tachycardia/hypertension**: propranolol or labetalol; magnesium. **Agitation/psychosis**: benzodiazepines, haloperidol/phenothiazines (avoid barbiturates). **Hyponatraemia**: fluid restrict if mild; **3 % hypertonic saline** if severe (Na <120) or seizing.
5. Monitor and treat complications
**Respiratory failure** from motor neuropathy is the chief killer — measure serial **FVC, MIP, MEP** (as in GBS); intubate early if FVC <15–20 mL/kg, declining trend, or bulbar weakness with aspiration. Treat SIADH hyponatraemia. Treat the precipitant (infection, etc.). Provide adequate **carbohydrate** nutrition enterally. Anticipate autonomic instability (arrhythmia, hypertension).
Drug safety in AIP — the exam table
The CICM/FFICM exam expects you to know which drugs are safe and which are dangerous. When in doubt, consult a current porphyria-safe drug list (these are updated regularly). [1]
❌ UNSAFE — AVOID (induce ALAS1)
High-risk precipitants
- Induction agents: **thiopentone**, barbiturates, etomidate.
- Anticonvulsants: phenytoin, carbamazepine, valproate, phenobarbitone, primidone.
- Antibiotics: sulfonamides, rifampicin/rifabutin, macrolides (variable), metronidazole (variable), griseofulvin.
- Hormones: combined oral contraceptives, progestogens, anabolic steroids, danazol.
- Others: alcohol, ergotamine, methyldopa, pentazocine, glutethimide.
✅ SAFE — USE FREELY
Porphyria-friendly
- Induction/sedation: **propofol**, ketamine, midazolam, other benzodiazepines, dexmedetomidine, volatile agents (sevoflurane/isoflurane), nitrous oxide.
- Analgesia: opioids — morphine, fentanyl, remifentanil, oxycodone, codeine; paracetamol; ketamine.
- Anticonvulsants: **gabapentin, pregabalin, levetiracetam**; benzodiazepines; magnesium sulphate.
- Antiemetics: ondansetron, prochlorperazine, domperidone.
- Cardiovascular: beta-blockers (propranolol, labetalol, esmolol), calcium-channel blockers, ACE inhibitors, nitrates, magnesium.
- Antibiotics: penicillins/cephalosporins, aminoglycosides, vancomycin, fluconazole.
- Neuromuscular blockers: suxamethonium, rocuronium, vecuronium, cisatracurium.
Worked example — a perioperative trap
Thiopentone for RSI in an undiagnosed carrier
Scenario
A 28-year-old woman is admitted with "acute abdomen" and agitation. RSI uses thiopentone. Within 48 h she develops ascending weakness, hypertension, tachycardia and Na 118.
What happened
She is an undiagnosed AIP carrier; thiopentone (a barbiturate) induced hepatic ALAS1 → ALA/PBG surge → neurovisceral attack compounded by SIADH.
Correct RSI in suspected/known AIP
Induce with **propofol** (or ketamine/etomidate-sparing regimen); avoid thiopentone entirely. Analgesia with fentanyl; paralysis with rocuronium/suxamethonium (all safe). Maintain anaesthesia with propofol or sevoflurane.
Take-home
ALWAYS include AIP in the differential of an atypical abdomen + neuropsychiatric signs, and default to **porphyria-safe drugs** (propofol-based) until the spot urinary PBG returns.
Complications and prognosis
Acute complications
During the attack
- Respiratory failure from motor neuropathy (the leading cause of death — monitor FVC).
- Seizures (primary porphyric or from hyponatraemia).
- Severe hyponatraemia → coma, cerebral oedema.
- Aspiration pneumonia from bulbar weakness / ileus.
- Hypertensive emergency, arrhythmias from autonomic storm.
- Hospital-acquired infection / line sepsis (central line for haem).
Chronic / long-term
Between and after attacks
- Chronic neuropathic pain, anxiety, depression and reduced quality of life even between attacks.
- Chronic kidney disease — from long-standing hypertension and possibly ALA nephrotoxicity; some therapies (givosiran) can worsen eGFR.
- Chronic hypertension, liver fibrosis/cirrhosis.
- **Hepatocellular carcinoma** — markedly increased risk; screen known AIP carriers with annual liver ultrasound ± AFP from age 50 (some guidelines earlier).
- Attack frequency is reduced by prophylaxis and trigger avoidance; severe recurrent disease can be transformed by givosiran or cured by liver transplant.
Prevention and long-term management
Preventing recurrence
1. Trigger avoidance (for everyone with AIP)
Lifelong porphyria-safe drug list; avoid crash diets and prolonged fasting (maintain regular carbohydrate intake); avoid alcohol; immunise against infections; plan surgery with a porphyria-aware team and minimise pre-op fasting.
2. Identify and treat the pattern of attacks
Document triggers and timing. Premenstrual/cyclical attacks → consider **GnRH analogue** (goserelin) with hormone add-back. Frequent non-cyclical attacks → consider prophylactic **haem arginate** (e.g. weekly) or **givosiran**.
3. Givosiran (Givlaari) — siRNA prophylaxis
Subcutaneous siRNA targeting hepatic ALAS1 (2.5 mg/kg monthly). ENVISION trial showed a **74 % reduction in annualised attack rate** vs placebo. Adverse effects: transaminitis (monitor LFTs), renal eGFR decline, injection-site reactions, fatigue, nausea. Reserved for recurrent attacks (≥3–4/year).
4. Definitive cure for severe refractory disease
**Liver transplantation** replaces the deficient hepatic HMBS and is curative for severe, treatment-refractory AIP; reserved for life-threatening, recurrent disease not controlled by haem/givosiran. Kidney transplant may be needed for end-stage renal disease.
5. Long-term surveillance
Annual blood pressure, renal function and LFTs; **annual hepatocellular carcinoma surveillance** (liver ultrasound ± AFP) in adult carriers. Genetic counselling and cascade testing of first-degree relatives.
Clinical presentation
Neurovisceral triad
Classic features
- Abdominal pain (#1 symptom): severe, colicky, diffuse, out of proportion to physical findings
- Neurological: motor neuropathy (proximal > distal, ascending — resembles GBS), seizures, psychiatric (anxiety, hallucination, paranoia), cranial neuropathies
- Autonomic: tachycardia, hypertension, constipation, urinary retention, sweating
- Hyponatraemia (SIADH from hypothalamic involvement)
Triggers
Precipitants
- Drugs: barbiturates (#1 trigger), sulfonamides, phenytoin, carbamazepine, oral contraceptives, ergotamines, rifampicin
- Alcohol, fasting/dieting, infection, surgery, stress
- Menstrual cycle (premenstrual attacks in women)
- Always check ALL medications against porphyria-safe drug list
Management
AIP acute attack management
Stop ALL triggering drugs
Review entire drug list. Stop: barbiturates, sulfonamides, phenytoin, carbamazepine, oral contraceptives, alcohol. Check ALL drugs against a porphyria-safe drug list (available from porphyria specialist centres). Replace with safe alternatives (opioids for pain, propofol for sedation, beta-blockers for hypertension).
IV hemin (haem arginate)
Hemin 3-4 mg/kg IV once daily for 4 days. Mechanism: exogenous haem represses ALA synthase (the rate-limiting enzyme that is upregulated in AIP) → reduces toxic precursor (ALA, PBG) production → reduces neurological damage. Give EARLY — delays increase severity and duration. Side effects: phlebitis (give via central line), anticoagulant effect, iron overload (rare with short courses).
IV glucose (if hemin unavailable or delayed)
10% dextrose infusion at 300-500 mL/h (or 300 g/day). Glucose inhibits ALA synthase (same mechanism as hemin but less potent). Useful as temporising measure while awaiting hemin, or in mild attacks. Monitor glucose (hyperglycaemia), sodium (SIADH is common).
Symptom control
Pain: opioids (morphine, fentanyl — safe in porphyria). Nausea: ondansetron (safe). Seizures: gabapentin, levetiracetam (safe — avoid phenytoin/carbamazepine/barbiturates). Hypertension/tachycardia: beta-blockers (propranolol — safe). Psychiatric: benzodiazepines (safe — avoid barbiturates).
Monitor for complications
Motor neuropathy: can progress to respiratory failure (like GBS). Monitor FVC, MIP, MEP. Intubate if FVC <15-20 mL/kg or bulbar weakness. Hyponatraemia: from SIADH — fluid restrict if mild, hypertonic saline if severe with seizures. Hypertension: treat with beta-blockers.
SAQ — Acute intermittent porphyria: diagnosis and management
SAQ — Acute intermittent porphyria presenting as an acute abdomen
10 minutes · 10 marks
A 26-year-old woman is admitted with severe, diffuse, colicky abdominal pain for 24 hours. She is tachycardic (HR 118), hypertensive (160/100), constipated and agitated. Her abdomen is soft and non-tender with no guarding. Na 118 mmol/L, normal lipase, normal CT abdomen. She had a similar episode 3 months ago that resolved spontaneously, and has a lower midline scar from a negative laparotomy during that admission. She started the combined oral contraceptive pill 6 weeks ago.
SAQ — AIP with ascending motor neuropathy, respiratory failure and SIADH
10 minutes · 10 marks
A 31-year-old woman with known AIP (HMBS mutation) is admitted with a severe acute attack precipitated by an intercurrent urinary tract infection. By day 3 she has developed progressive ascending weakness — now unable to lift her arms off the bed, with reduced reflexes and FVC of 17 mL/kg (falling). Her sodium is 116 mmol/L. She is agitated and has had one generalised seizure.
Clinical pearls
Trial card
ENVISION — Givosiran for Acute Intermittent Porphyria
PMID 32521132
Population: 94 patients with AIP and ≥2 attacks in the prior 6 months (or ≥4/year on prophylaxis requiring hospitalisation, urgent visit, or IV haem).
Key finding
Annualised attack rate reduced from 10.7 (placebo) to 3.2 (givosiran) — **rate ratio 0.26, a ~74 % reduction** (p<0.001). Significant reductions in urinary ALA and PBG, haem use, daily pain and quality-of-life scores.
Red flags
Exam mnemonics
- "4 × PBG → HMB" — PBG deaminase condenses 4 molecules of PBG into hydroxymethylbilane (the AIP block).
- "5 Ds of triggers": Drugs, Diet (fasting), Drink (alcohol), Disease (infection/surgery), Diary (menstrual).
- "AIP = Abdomen + Insomnia/Insanity + Peripheral (motor) neuropathy" — the triad.
- "Givosiran = ALAS1 silencer (G for Genes off)" — siRNA → ↓ ALA/PBG → fewer attacks.
- "Glucose guards ALAS1" — carbohydrate represses ALA synthase (the glucose effect).
- "Porphyrin pearls: AIP=abdomen only; PCT=Photoskin; VP=Variety (skin + gut)" — skin involvement distinguishes them. [1]
One-page summary
AIP is an autosomal-dominant, low-penetrance defect of HMBS (PBG deaminase), the third enzyme of haem synthesis. Attacks occur only when hepatic ALAS1 is induced — by barbiturates, antiepileptics, sulfonamides, rifampicin, hormones, alcohol, fasting, infection or stress — driving accumulation of the neurotoxic precursors ALA and PBG. The presentation is a neurovisceral triad of severe abdominal pain (benign abdomen), motor neuropathy / seizures / psychiatric change, and autonomic storm (tachycardia, hypertension, constipation), frequently with SIADH hyponatraemia. Diagnosis rests on a markedly elevated random spot urinary PBG (with ALA), differentiation by faecal porphyrins and confirmation by HMBS sequencing. Treatment is withdrawal of triggers, IV haem arginate 3 mg/kg daily × 4 days (glucose ~300 g/day if unavailable), opiate analgesia, phenothiazines/ondansetron for nausea, gabapentin/levetiracetam for seizures, correction of hyponatraemia, and respiratory monitoring for motor neuropathy. Prevention relies on trigger avoidance, GnRH analogues for cyclical disease, givosiran (ENVISION, NEJM 2020) for frequent attacks, and liver transplant for refractory disease, with lifelong surveillance for chronic kidney disease and hepatocellular carcinoma. [1]
References
- [1]Besur S, Schmeltzer P, Bonkovsky HL. Acute Porphyrias J Emerg Med, 2015.PMID 26159905
- [2]Bissell DM, Anderson KE, Bonkovsky HL. Porphyria N Engl J Med, 2017.PMID 28854095
- [3]Besur S, Hou W, Schmeltzer P, Bonkovsky HL. Clinically important features of porphyrin and heme metabolism and the porphyrias Metabolites, 2014.PMID 25372274
- [4]Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria Appl Clin Genet, 2015.PMID 26366103
- [5]Pischik E, Kauppinen R. Neurological manifestations of acute intermittent porphyria Cell Mol Biol (Noisy-le-grand), 2009.PMID 19268005
- [6]Balwani M, Sardh E, Ventura P, et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria N Engl J Med, 2020.PMID 32521132
- [7]Syed YY. Givosiran: A Review in Acute Hepatic Porphyria Drugs, 2021.PMID 33871817