ICU · Neurocritical Care
Myasthenia gravis crisis and neuromuscular respiratory failure
Also known as Myasthenic crisis · Cholinergic crisis · Neuromuscular respiratory failure · Plasmapheresis · IVIG for myasthenia
Myasthenic crisis is respiratory failure from severe weakness of respiratory muscles (diaphragm, intercostals) and bulbar muscles (swallowing, airway protection) in myasthenia gravis, an autoimmune disorder of the postsynaptic neuromuscular junction. Antibodies target the acetylcholine receptor (AChR, ~85% of generalised MG), muscle-specific tyrosine kinase (MuSK, 5-8%), or LRP4. Triggers: infection (1), surgery, pregnancy and the postpartum period, and drugs that impair neuromuscular transmission (aminoglycosides, magnesium, beta-blockers, fluoroquinolones, neuromuscular blockers, corticosteroid initiation). Presents with progressive fatigable weakness, dysphagia, dysarthria, weak cough, and respiratory distress. Diagnosis is clinical (known MG with worsening fatigable weakness) supported by bedside tests (fatigability, ice pack test, single breath count) and bedside pulmonary function (FVC, MIP, MEP), and confirmed by AChR/MuSK antibodies, repetitive nerve stimulation and single-fibre EMG. Management: ICU admission for respiratory monitoring, ventilatory support (NIV first, then intubation if bulbar/respiratory failure), IVIG or plasma exchange (equally effective), identify and treat the trigger, and start or adjust immunosuppression (prednisolone with an early-steroid-worsening caveat, azathioprine, mycophenolate, rituximab). Thymectomy is mandatory for thymoma and beneficial in selected AChR-positive generalised MG. Distinguish myasthenic crisis (under-treated, dry) from cholinergic crisis (over-treated with anticholinesterase, wet — SLUDGE). Mortality 4-8% with modern ICU care.
On this page & tools
Your progress
Saved locally on this device.
Target exams
Red flags

Pathophysiology — the disease at the junction

Myasthenia gravis is a T-cell-dependent, antibody-mediated, postsynaptic disorder of the neuromuscular junction (NMJ). The motor nerve terminal releases acetylcholine (ACh) into the synaptic cleft; ACh binds nicotinic ACh receptors (AChR) on the motor end-plate, depolarising the muscle fibre. In MG, pathogenic autoantibodies attack the postsynaptic membrane, reducing the available receptor reserve. Because the normal junction has a large safety factor (a single nerve impulse releases far more ACh than is needed to trigger contraction), the patient is asymptomatic at rest. As transmission fatigues with sustained effort, weakness emerges — the hallmark fatigability. The respiratory and bulbar muscles, which must sustain high-frequency firing, are the first to fail in crisis. [1]
The antibody target defines the phenotype, the response to treatment, and the prognosis. [1]
Antibody profile in generalised myasthenia gravis
AChR-positive MG
- Most common subtype (~85%); IgG1/IgG3, fixes complement
- Thymic abnormalities common — thymoma in ~10-15%, follicular thymic hyperplasia in ~65%
- Ocular onset common, generalises in most; classic fatigable limb, bulbar, respiratory weakness
- Good response to acetylcholinesterase inhibitors, corticosteroids, azathioprine, thymectomy (if non-thymomatous, AChR+ generalised)
MuSK-positive MG
- ~5-8%; IgG4 (poor complement fixation) against muscle-specific tyrosine kinase
- Predominantly bulbar, facial, and respiratory weakness; tongue and muscle atrophy may occur; little ocular involvement
- Female predominance, onset typically <40 years; thymus usually normal — thymectomy NOT indicated
- Poor response to acetylcholinesterase inhibitors (can worsen); excellent response to plasmapheresis and rituximab; prone to crisis
LRP4-positive MG
- ~2-5%; antibody against LRP4 in the agrin–MuSK AChR-clustering pathway
- Usually milder phenotype, frequent ocular involvement, less respiratory failure
- Lower crisis rate; responds to standard immunosuppression
- Often grouped with seronegative disease on routine panels
Seronegative MG
- ~5%; antibodies not detected by standard cell-based assays
- Many have antibodies against AChR clustered on the cell surface or low-affinity IgG
- Treat as AChR-positive MG clinically
- Specialised reference laboratories clarify most cases
The distinction matters at the bedside: MuSK MG tends to present in crisis with severe bulbar and respiratory weakness, responds poorly to pyridostigmine (which may even worsen it) and very well to plasmapheresis and rituximab, and is not helped by thymectomy. AChR MG is the prototype that drives the monitoring thresholds, the immunosuppression ladder, and the thymectomy decision.[2]
Triggers of myasthenic crisis
Crisis rarely arises de novo — it is almost always precipitated. Identifying and treating the trigger is as important as treating the weakness, because failure to remove the precipitant predicts relapse and prolonged ventilation. In a typical series, infection accounts for around 30-40% of crises, aspiration and surgery each around 15-20%, and medication change or immunosuppression taper another 15%; a substantial minority remain idiopathic.[1][7]
Search for and treat the precipitant
Infection (the single most common trigger, ~30-40%)
Lower respiratory tract infection and pneumonia lead. Send chest X-ray, sputum, urine and blood cultures on admission. Treat with MG-safe antibiotics — cephalosporins, penicillins, macrolides (caution), carbapenems, linezolid, vancomycin. AVOID aminoglycosides (gentamicin, tobramycin, amikacin — presynaptic calcium blockade reduces ACh release), and use fluoroquinolones, telithromycin and polymyxins only if no alternative.
Aspiration and surgery
Aspiration pneumonitis from bulbar weakness both triggers and complicates crisis. Perioperative crisis is common after thymectomy and other major surgery — the stress response, anaesthetic agents and neuromuscular blockers all conspire. Anticipate postoperative respiratory monitoring; elective surgery should be done when MG is well controlled, with the morning pyridostigmine dose given.
Pregnancy and the postpartum period
MG often improves in the second trimester and worsens in the first trimester, the puerperium (first 30 days postpartum) and with the onset of lactation. Pre-eclampsia treatment with magnesium sulfate is catastrophic in MG. Plan delivery in a unit with neurology, obstetrics and ICU; transient neonatal MG occurs in ~10-20% of babies from transplacental AChR antibody transfer.
Medication change (drugs that impair NMJ transmission)
Review every drug against an MG-safe list. The high-yield offenders: aminoglycosides, magnesium, beta-blockers (including timolol eye drops), calcium channel blockers, fluoroquinolones, macrolides, neuromuscular blockers, iodinated contrast, telithromycin and botulinum toxin. Steroid initiation is a special case — see below.
Tapering immunosuppression and emotional/physical stress
A recent reduction in prednisolone, azathioprine or other immunosuppressant is a classic precipitant. Heat, systemic illness, surgery, emotional stress and thyroid dysfunction (hyper- or hypothyroidism) also lower the safety factor. Check thyroid function on every admission.
Definitely avoid
- Aminoglycosides — gentamicin, tobramycin, amikacin, neomycin
- Magnesium sulfate (including pre-eclampsia/eclampsia dosing)
- Botulinum toxin — worsens weakness
- Neuromuscular blocking agents — prolonged paralysis in MG
- Telithromycin
Use with caution
- Fluoroquinolones (ciprofloxacin, levofloxacin)
- Macrolides (azithromycin, clarithromycin, erythromycin)
- Beta-blockers including timolol eye drops
- Calcium channel blockers
- Iodinated contrast media
- Corticosteroids on initiation (transient worsening)
Generally safe
- Penicillins and cephalosporins
- Carbapenems (meropenem, imipenem)
- Vancomycin, linezolid, teicoplanin
- Azole and echinocandin antifungals
- Paracetamol/acetaminophen, opioids for analgesia
- Propofol, volatile agents, fentanyl for anaesthesia
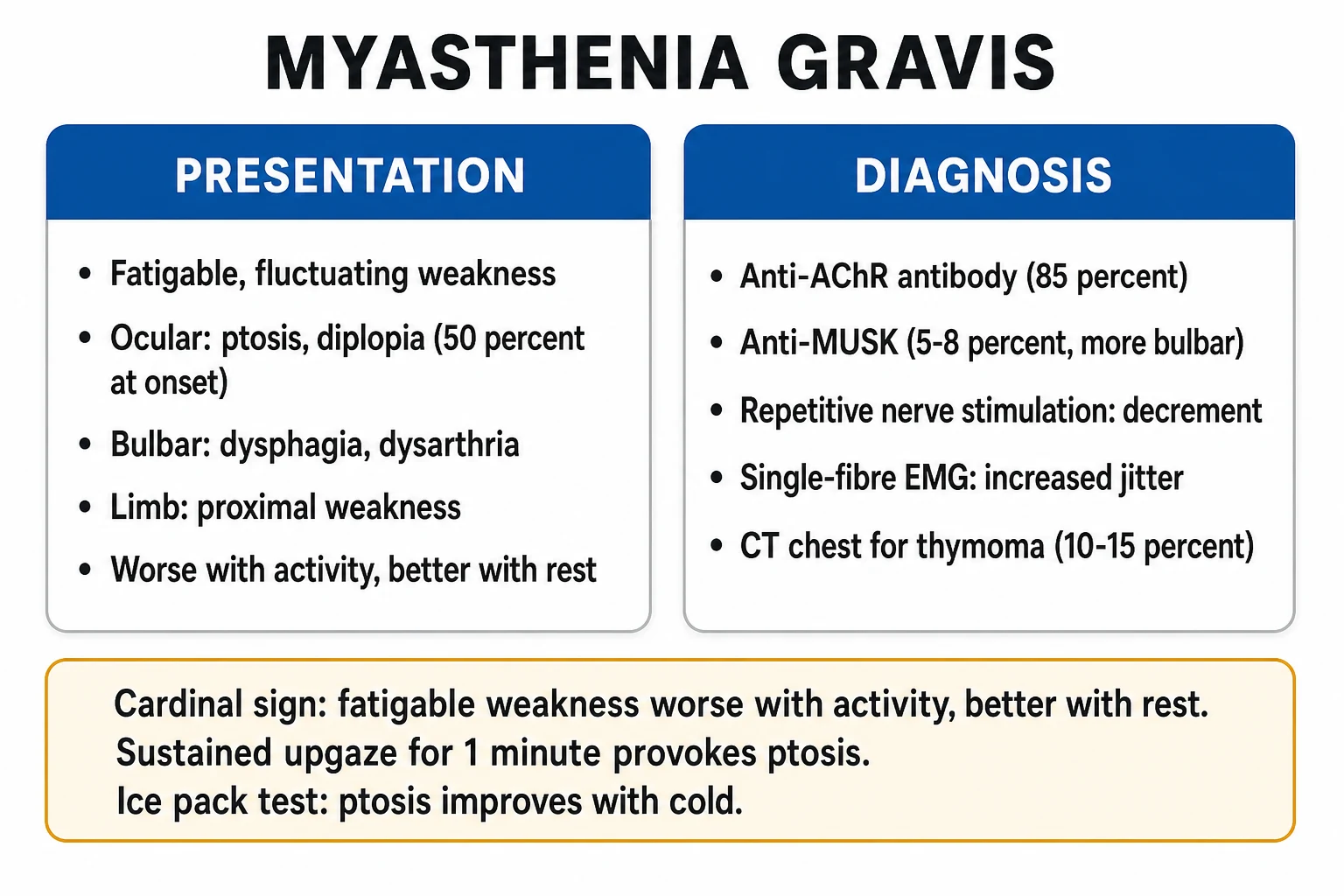
Clinical presentation and bedside diagnosis
The crisis is a clinical diagnosis made on the background of known (or occasionally newly presenting) MG. Weakness is fatigable — worse with sustained or repeated effort, better with rest — and progresses over hours to days. The danger signs are respiratory and bulbar. [1]
Bedside assessment in suspected crisis
Demonstrate fatigability
Sustained upgaze for 60-120 s produces ptosis; repeated arm abduction or rising from a chair 20 times causes proximal weakness; counting out loud reveals a fading, nasal dysarthria. Fatigability that recovers with a minute of rest is the clinical fingerprint of the postsynaptic block.
Single breath count (SBC)
Ask the patient to count aloud at one number per second after a maximal inspiration on a single breath. A count <30 suggests FVC around 1.5-2 L; <20 suggests impending failure. The SBC is a rapid, equipment-free trend monitor — a falling trend over hours is an early warning to intubate.
Ice pack test
A 2-minute application of ice in a glove to a ptotic eyelid reduces acetylcholinesterase activity locally and improves myasthenic ptosis in most AChR-positive patients (sensitivity ~90%). Simple, non-invasive, useful in the undiagnosed ptotic patient. Largely a screening test — it does not quantify respiratory risk.
Bulbar assessment
Assess speech (nasal or hoarse, fading voice), swallow (cough on thin fluids, pooled secretions), and cough strength (weak cough cannot clear secretions). Bulbar weakness is an independent indication for intubation because of aspiration risk, even with acceptable FVC.
Serial bedside pulmonary function
Measure FVC, MIP (maximum inspiratory pressure, = NIF) and MEP (maximum expiratory pressure) every shift and with any deterioration. FVC <15-20 mL/kg (or <1 L), MIP less negative than -30 cmH2O, or MEP <40 cmH2O predict ventilatory failure. The trend matters more than any single value.
Confirmatory investigations
Bedside tests establish the crisis; laboratory tests confirm the underlying MG and exclude mimics. In a patient with established MG, treatment must not be delayed while awaiting antibody results — they take days. [1]
Confirming myasthenia gravis
Antibody testing
AChR-binding antibodies are positive in ~85% of generalised MG (sensitive and specific). MuSK antibodies are tested when AChR is negative, positive in 5-8% (higher in pure bulbar/respiratory phenotypes). LRP4 antibodies on specialist assays clarify some seronegative cases. Antibody titre does not correlate closely with severity and should not guide acute dosing.
Repetitive nerve stimulation (RNS)
A decremental response (>10% amplitude fall) on low-frequency (3 Hz) stimulation of a proximal or facial nerve is characteristic. Sensitivity rises with the number of muscles tested (spinal accessory, facial, proximal limb). A normal RNS does not exclude MG.
Single-fibre EMG
The most sensitive test (>95% in generalised MG): it shows increased jitter and blocking. Highly sensitive but non-specific — abnormal in any disorder that impairs neuromuscular transmission, so it is used to confirm, not to define aetiology.
CT or MRI of the thymus
Perform in all newly diagnosed MG patients. Thymoma (~10-15% of AChR-positive MG) mandates surgical resection. Thymic hyperplasia is common and is not itself an indication for surgery.
Exclude mimics and check thyroid
Thyroid function (dysthyroid eye disease, and thyroid dysfunction worsens MG), creatine kinase (myositis), B12, and — if acute and asymmetric — imaging to exclude stroke. If acute ascending paralysis is present, consider Guillain-Barre syndrome instead.
AChR antibodies
- ~85% of generalised MG; highly specific
- Positive result confirms MG; titre does not track severity
- Test first-line in any suspected case
- Negative in most pure ocular and MuSK cases
Repetitive nerve stimulation
- 3 Hz stimulation; >10% decrement is positive
- Sensitivity ~75-85% in generalised MG if multiple muscles tested
- Normal in some MuSK MG
- Bedside-accessible in many ICUs
Single-fibre EMG
- Most sensitive test (>95%)
- Shows increased jitter and impulse blocking
- Non-specific — abnormal in any NMJ disorder
- Confirms a disorder of transmission, not the cause
Edrophonium (Tensilon) test
- Historical bedside diagnostic test
- Largely abandoned — risk of bradycardia, bronchospasm, syncope
- Replaced by antibody testing and ice pack test
- Not used in the modern ICU assessment
Monitoring
Myasthenic crisis monitoring protocol
Serial pulmonary function
Monitor EVERY SHIFT: FVC (forced vital capacity), MIP (maximum inspiratory pressure, also called NIF — negative inspiratory force), MEP (maximum expiratory pressure). Trends are more important than absolute values. Intubation criteria: FVC <15-20 mL/kg (or rapidly declining), MIP < -30 cmH2O (less negative = weaker), MEP < 40 cmH2O. Bulbar weakness (dysphagia, weak cough, secretion pooling) is an independent indication for intubation (aspiration risk).
Avoid intubation if possible
NIV (BiPAP) may prevent intubation in selected patients (cooperative, able to protect airway, no severe bulbar weakness). NIV reduces work of breathing, rests fatigued muscles. BUT: if bulbar weakness present → aspiration risk → intubate. Do NOT delay intubation — elective intubation is safer than emergency.
Ventilation strategy
If intubated: standard lung-protective ventilation. Most MG patients have normal lungs — ventilation should be straightforward. Wean as MG improves (IVIG/plasmapheresis takes 1-2 weeks). Extubation criteria: FVC >15 mL/kg, MIP > -30 cmH2O, MEP > 40 cmH2O, adequate cough, minimal secretions.
Treatment
Myasthenic crisis treatment
IVIG or plasmapheresis
IVIG 0.4 g/kg/day x 5 days OR plasmapheresis (5 sessions over 1-2 weeks). Equally effective (no trial has shown superiority of either). Choice based on: availability, comorbidities (plasmapheresis needs central line + bleeding risk; IVIG needs venous access + thrombosis risk). Effect: removes/blocks acetylcholine receptor antibodies → improves neuromuscular transmission. Onset: 1-2 weeks. Duration: weeks-months.
Corticosteroids
Prednisolone 1-1.5 mg/kg/day. CAUTION: steroids may initially WORSEN myasthenia (first 5-10 days) — monitor closely, consider starting in ICU or with concurrent IVIG/plasmapheresis. Steroids reduce antibody production over weeks-months. Taper to lowest effective dose. Most MG patients need long-term immunosuppression.
Identify and treat trigger
Search for: (1) Infection (#1 trigger — chest X-ray, urine, blood cultures, treat with MG-safe antibiotics). (2) Surgery (thymectomy — may trigger crisis postoperatively). (3) Pregnancy. (4) Drug change (new medication that worsens MG — check ALL drugs against MG-safe list). (5) Immunization taper (reduced immunosuppression → flare).
Continue/adjust pyridostigmine
Pyridostigmine (acetylcholinesterase inhibitor): continue orally/NG if possible (symptomatic treatment — does NOT modify disease). Reduce dose if cholinergic side effects (excess salivation, diarrhoea, bradycardia). IV pyridostigmine: 1/30th of oral dose (proportional — much smaller IV dose). Discontinue temporarily if intubated and on IVIG/plasmapheresis (these will improve the underlying disease — anticholinesterase may not be needed acutely).
Thymectomy evaluation
If thymoma present: surgical resection (usually after crisis resolves — not during acute crisis). If no thymoma: thymectomy may benefit patients with acetylcholine receptor antibody-positive generalized MG (MGTX trial — improved outcomes over 3 years). Timing: after crisis resolution (surgery itself can trigger crisis).
IVIG (intravenous immunoglobulin)
Short-term immunomodulation for acute myasthenic crisis
Dose (70kg)
28.0–70.0 g/day
Prednisolone
First-line long-term immunosuppression
Dose (70kg)
35.0–105.0 mg/day
Respiratory support — NIV first, then intubation
The respiratory muscles fail because the fatigued NMJ cannot sustain the high-frequency firing the diaphragm and intercostals require. The aim is to rest the fatigued junction while IVIG or plasma exchange takes effect, intubating before the patient arrests. [1]
Airway and ventilation ladder
Trial of NIV (BiPAP)
For the cooperative patient WITHOUT severe bulbar weakness, BiPAP (IPAP 10-14, EPAP 4-6, titrated) reduces the work of breathing and rests fatigued muscles, and can avoid intubation in selected cases. Contraindicated by bulbar weakness, inability to protect the airway, secretion pooling, or reduced conscious state — all mandate intubation rather than NIV.
Early elective intubation
Intubate for FVC <15-20 mL/kg (or a rapid downward trend), MIP less negative than -30 cmH2O, MEP <40 cmH2O, single breath count <20, severe bulbar weakness, aspiration, or fatigue. Elective, planned intubation is far safer than an emergency one. Use a lung-protective ventilatory strategy; most MG patients have normal lungs.
Neuromuscular blockade — avoid if possible
MG patients are exquisitely sensitive to non-depolarising neuromuscular blockers (prolonged paralysis) and relatively resistant to suxamethonium (higher doses needed, risk of phase II block). For RSI use a reduced dose of rocuronium (0.6 mg/kg) or vecuronium with train-of-four monitoring, or avoid NMBA altogether with a high-dose opioid/volatile technique. Sugammadex reversal is valuable.
Weaning and extubation
Wean as IVIG/PLEX takes effect (typically 1-2 weeks). Extubate when FVC >15 mL/kg, MIP more negative than -30 cmH2O, MEP >40 cmH2O, strong cough, minimal secretions, and bulbar function recovered. A cuff-leak test and a spontaneous breathing trial help. Tracheostomy is reserved for prolonged failure (>2-3 weeks) — most patients wean as the immunotherapy works.
Respiratory severity by bedside thresholds
Severe
FVC <15 mL/kg or single breath count <20, MIP less negative than -30 cmH2O, MEP <40 cmH2O, or severe bulbar weakness with secretion pooling. Intubate electively.
Myasthenic vs cholinergic crisis — the exam-defining distinction
This is the most commonly examined distinction in MG crisis, because the treatments are opposite. Both produce weakness; the rest of the picture tells them apart. The Tensilon (edrophonium) test is now avoided because it can precipitate bradycardia, bronchospasm and worse weakness if the diagnosis is wrong — but historically a brief improvement pointed to myasthenic crisis. [1]
Myasthenic crisis
- UNDER-treated MG — too little anticholinesterase or a new trigger
- Weakness WITHOUT cholinergic excess
- Dry: dry mouth, dry skin, mydriasis (pupil), tachycardia
- NO fasciculations; reduced/absent reflexes from weakness
- Improves with edrophonium (historically); treat with IVIG/PLEX and increase immunosuppression
Cholinergic crisis
- OVER-treated — excess acetylcholinesterase inhibitor (pyridostigmine/neostigmine)
- Weakness WITH cholinergic nicotinic and muscarinic excess
- Wet — SLUDGE: Salivation, Lacrimation, Urination, Diaphoeesis/Diarrhoea, GI cramps, Emesis
- Miosis, bradycardia, and fasciculations (nicotinic overdrive)
- Treat by stopping anticholinesterase, atropine for muscarinic effects, supportive ventilation; may need IVIG/PLEX if mixed
How to tell at the bedside
- Examine for SLUDGE and the pupils/heart rate: wet, miosis, bradycardia = cholinergic; dry, mydriasis, tachycardia = myasthenic
- Fasciculations strongly favour cholinergic (nicotinic) excess
- A recent large pyridostigmine dose increase or IV neostigmine points to cholinergic
- If genuinely unclear, treat as myasthenic (the more dangerous default) and withdraw anticholinesterase temporarily while reassessing
Immunotherapy and thymectomy
Beyond the acute short-term therapy (IVIG/PLEX), most MG patients need long-term immunosuppression to maintain remission, and selected patients benefit from thymectomy. Steroids are first-line but have an early-worsening caveat; steroid-sparing agents take months to work. [1]
Long-term immunosuppression ladder
Pyridostigmine (symptomatic only)
Acetylcholinesterase inhibitor (60 mg orally, typically q6h). Symptomatic — it does NOT modify the underlying disease. Useful in mild AChR MG; often unhelpful or worsening in MuSK MG. Taper or hold in intubated crisis.
Corticosteroids (first-line immunosuppressant)
Prednisolone 0.5-1.5 mg/kg/day. Effective and fast (2-4 weeks), but the early-steroid-worsening phenomenon mandates monitored initiation, often with concurrent IVIG/PLEX. Taper to the lowest effective maintenance dose and add a steroid-sparing agent early.
Steroid-sparing oral agents
Azathioprine (1-3 mg/kg/day; first-line steroid-sparer, 6-12 months to effect, check TPMT), mycophenolate mofetil (2 g/day, 3-6 months to effect), methotrexate, tacrolimus and cyclosporine. These allow prednisolone tapering and sustain remission.
Targeted biologic therapy
Rituximab (anti-CD20) is highly effective, especially in refractory and MuSK MG. Eculizumab (terminal complement inhibitor) and ravulizumab are approved for refractory AChR-positive generalised MG (REGAIN trial). Used in refractory disease or as rapid steroid-sparing therapy.
Thymectomy
Mandatory for thymoma (all grades). In non-thymomatous AChR-positive generalised MG aged 18-65, the MGTX trial showed thymectomy plus prednisolone improved outcomes and allowed lower steroid doses over 3 years. NOT indicated in MuSK MG (normal thymus, no benefit).
Prednisolone
- First-line immunosuppressant; fast onset (2-4 weeks)
- Can WORSEN MG in first 5-10 days — start monitored or with IVIG/PLEX
- Long-term toxicity: diabetes, osteoporosis, hypertension, infection
- Always add a steroid-sparing agent early
Azathioprine
- First-line steroid-sparer; check TPMT before starting
- Slow onset — 6-12 months to full effect
- Monitor FBC and LFTs; hepatotoxicity and myelosuppression
- Allows prednisolone tapering; widely used first-line
Mycophenolate mofetil
- Alternative steroid-sparer; 3-6 months to effect
- GI upset, myelosuppression, teratogenic (avoid in pregnancy)
- Widely used; some trial evidence is mixed for induction but good for maintenance
- Favoured where azathioprine is not tolerated
Rituximab
- Anti-CD20 B-cell depletion; dramatic in MuSK and refractory AChR MG
- BeatMG phase 2 in AChR MG did not meet its primary endpoint but rituximab is widely used off-label
- Infusion reactions, infection, reactivation of hepatitis B
- Increasingly first biologic in MuSK MG and refractory disease
Differential diagnosis of acute neuromuscular respiratory failure
Not every patient with weak breathing has myasthenia. The pattern of weakness, the reflexes and the time course separate the NMJ disorders from pre- and post-synaptic mimics. [1]
Myasthenia gravis (crisis)
- Postsynaptic; fatigable weakness, ocular/bulbar prominent
- Reflexes usually preserved (reduced only by weakness)
- Sensory examination normal; no autonomic failure beyond bulbar
- AChR/MuSK antibodies; RNS decrement; responds to IVIG/PLEX
Guillain-Barre syndrome
- Acute ascending flaccid paralysis with areflexia
- Preceded by infection (Campylobacter); days to nadir
- Sensory loss and autonomic instability common
- Nerve conduction: demyelination; treat with IVIG or plasma exchange
Lambert-Eaton myasthenic syndrome
- Presynaptic; antibodies against voltage-gated calcium channels
- Weakness IMPROVES with repeated activity (facilitation)
- Autonomic dysfunction and areflexia; strong paraneoplastic link (small-cell lung cancer)
- RNS shows incremental response; treat the tumour, amifampridine, IVIG
ICU-acquired weakness (CIP/CIM)
- Develops AFTER days in ICU (sepsis, steroids, immobility)
- Diffuse limb weakness, difficult weaning, reduced reflexes
- Not present on admission — distinguishes from MG crisis
- Diagnosis by electrophysiology; prevention is key
Landmark trials
MGTX — Thymectomy in non-thymomatous AChR-positive generalised MG
New England Journal of Medicine, 2016
A randomised, rater-blinded trial of extended transsternal thymectomy plus prednisolone versus prednisolone alone in 126 adults (18-65 years) with AChR-antibody-positive, non-thymomatous generalised myasthenia gravis of less than 5 years duration, followed for 3 years.
Key finding
Thymectomy plus prednisolone was superior to prednisolone alone across all primary endpoints: lower quantitative myasthenia gravis scores, lower prednisolone requirement, fewer hospitalisations, and more patients in minimal-manifestation state. The benefit persisted at extended follow-up, with prednisone withdrawal possible in a substantial proportion.
Practice change
Established thymectomy as standard for selected non-thymomatous AChR-positive generalised MG aged 18-65, performed to improve disease control and reduce long-term steroid burden. Not indicated in MuSK MG (normal thymus).
Gajdos — IVIG for myasthenia gravis (Cochrane)
Cochrane Database of Systematic Reviews, 2012
A systematic review and meta-analysis of randomised trials comparing intravenous immunoglobulin with placebo or with plasma exchange in myasthenia gravis, including acute exacerbation and crisis.
Key finding
IVIG was no different from plasma exchange in improving weakness in acute exacerbation, and both were superior to placebo. No single short-term therapy emerged as clearly superior; the two were considered equivalent in efficacy and safety in the acute setting.
Practice change
Codified IVIG and plasma exchange as equivalent short-term therapies for myasthenic crisis — the choice is made on availability and patient comorbidities (central access and bleeding for PLEX; thrombosis and renal risk for IVIG), not on efficacy.
BeatMG — Rituximab in AChR-positive generalised MG
Neurology, 2022
A phase 2, randomised, double-blind, placebo-controlled trial of rituximab (anti-CD20) versus placebo in 62 adults with AChR-antibody-positive generalised myasthenia gravis, with the primary endpoint of steroid-sparing effect.
Key finding
The trial did not meet its primary prespecified steroid-sparing endpoint in this AChR-positive population, although trends favoured rituximab and the drug was well tolerated. Retrospective data and clinical experience, especially in MuSK MG, continue to support marked benefit.
Practice change
Rituximab remains widely used off-label, particularly in refractory and MuSK MG where it is considered highly effective; the AChR-specific evidence is less definitive than the real-world experience in MuSK disease.
REGAIN — Eculizumab in refractory AChR-positive generalised MG
Neurology, 2021 (extension)
A phase 3 randomised, double-blind, placebo-controlled trial of eculizumab (terminal complement C5 inhibitor) in refractory AChR-antibody-positive generalised myasthenia gravis, with an open-label extension reporting post-intervention status.
Key finding
Eculizumab produced sustained improvements in activities of daily living, quantitative MG scores and myasthenia gravis activities of daily living over the extension, with benefits maintained across muscle groups. Requires meningococcal vaccination (overwhelming meningococcal infection risk).
Practice change
Established terminal complement inhibition as a targeted therapy for refractory AChR-positive MG, used when standard immunosuppression fails.
SAQ — Guillain-Barré syndrome with impending respiratory failure
10 minutes · 10 marks
A 52-year-old man presents with progressive ascending weakness over 5 days, beginning in his feet. He had a diarrhoeal illness (Campylobacter) two weeks ago. Examination: MRC sum score 30, flaccid limb weakness (power 3/5 in the legs, 4/5 in the arms), areflexia, bilateral facial weakness, and a weak cough with pooled secretions. FVC 18 mL/kg (falling from 25 mL/kg two hours ago), MIP -28 cmH2O, MEP 35 cmH2O. HR fluctuates between 45 and 110; BP swings from 90/50 to 170/95. ABG on room air: pH 7.32, PaCO2 48 mmHg, PaO2 78 mmHg, HCO3 26 mmol/L.
SAQ — Myasthenic crisis: airway decision and immunotherapy
10 minutes · 10 marks
A 38-year-old woman with known acetylcholine-receptor-antibody-positive myasthenia gravis, taking pyridostigmine 60 mg q6h and azathioprine 150 mg/day, presents after three days of worsening dysphagia, nasal speech and dyspnoea following a respiratory tract infection treated with azithromycin by her GP. She is diaphoretic and distressed, has ptosis on sustained upgaze, a fading nasal voice when counting, a weak cough with pooled secretions, and fatigable proximal limb weakness. FVC 16 mL/kg (down from 22 mL/kg yesterday), MIP -32 cmH2O, MEP 38 cmH2O. BP 145/88, HR 95, SpO2 94% on room air. No salivation, miosis, bradycardia, diarrhoea or fasciculations are present.
Clinical pearls
Red flags
Regional practice
ANZ practice note. Management follows the international consensus guidance and the MGFT/MGTX framework via local neurology and intensive-care protocols. Crisis is treated with IVIG 0.4 g/kg/day for 5 days or plasma exchange (5 sessions over 1-2 weeks), chosen by comorbidity and access; prednisolone is started under that cover and a steroid-sparing agent (azathioprine after TPMT, or mycophenolate) is introduced early. Bedside FVC/MIP/MEP thresholds (FVC <15-20 mL/kg, MIP < -30 cmH2O, MEP <40 cmH2O) drive the intubation decision, with bulbar weakness an independent trigger. Thymectomy is offered to thymoma patients and to selected non-thymomatous AChR-positive generalised MG patients (MGTX), performed after crisis resolution. Eculizumab and rituximab are accessed via neurology for refractory disease. [1]
References
- [1]Lacomis D. Myasthenic crisis Neurocrit Care, 2005.PMID 16377829
- [2]Gilhus NE. Myasthenia Gravis N Engl J Med, 2016.PMID 28029925
- [3]Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized Trial of Thymectomy in Myasthenia Gravis N Engl J Med, 2016.PMID 27509100
- [4]Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: Executive summary Neurology, 2016.PMID 27358333
- [5]Gajdos P, Chevret S, Toyka KV. Intravenous immunoglobulin for myasthenia gravis Cochrane Database Syst Rev, 2012.PMID 23235588
- [6]Nowak RJ, Coffey CS, Goldstein JM, et al. Phase 2 Trial of Rituximab in Acetylcholine Receptor Antibody-Positive Generalized Myasthenia Gravis: The BeatMG Study Neurology, 2022.PMID 34857535
- [7]Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis South Med J, 2008.PMID 18176295
- [8]Chaudhuri A, Behan PO. Myasthenic crisis QJM, 2009.PMID 19060020
- [9]Mandawat A, Kaminski HJ, Cutter G, Katirji B. Comparative analysis of therapeutic options used for myasthenia gravis Ann Neurol, 2010.PMID 21061395
- [10]Lee I, Kuo HC, Aban IB, Cutter GR. Minimal manifestation status and prednisone withdrawal in the MGTX trial Neurology, 2020.PMID 32611638
- [11]Mantegazza R, Wolfe GI, Muppidi S, et al. Post-intervention Status in Patients With Refractory Myasthenia Gravis Treated With Eculizumab During REGAIN and Its Open-Label Extension Neurology, 2021.PMID 33229455
- [12]Myllynen C, Tuulasvaara A, Atula S, Laakso SM. Intensive care due to myasthenia gravis: Risk factors and prognosis Eur J Neurol, 2024.PMID 39435628