ICU · Respiratory / gas exchange
Hypercapnia & Ventilatory (Type-2) Respiratory Failure
Also known as Hypercapnia · Type 2 respiratory failure · Ventilatory failure · Carbon dioxide retention · CO2 narcosis · Hypercapnic respiratory failure · Haldane effect · Oxygen-induced hypercapnia · Respiratory acidosis
Hypercapnia (a raised PaCO2) is ventilatory, or type-2, respiratory failure. Because PaCO2 is proportional to CO2 production divided by alveolar ventilation, hypercapnia arises from increased CO2 production (fever, sepsis, overfeeding) or — far more commonly — reduced alveolar ventilation from a failure of the controller (opioids, brainstem), the pump and nerves (Guillain-Barre, myasthenia), the chest wall and load (obesity, kyphoscoliosis, COPD, asthma), or increased dead space. The effects are respiratory acidosis, cerebral vasodilation with raised intracranial pressure (headache, asterixis, drowsiness, CO2 narcosis), and sympathetic stimulation. Treatment is to ventilate the patient (non-invasive ventilation for COPD) and reverse the cause; controlled oxygen in COPD averts oxygen-induced hypercapnia (the Haldane effect and worsened V/Q mismatch).
On this page & tools
Your progress
Saved locally on this device.
Target exams
Overview & definition
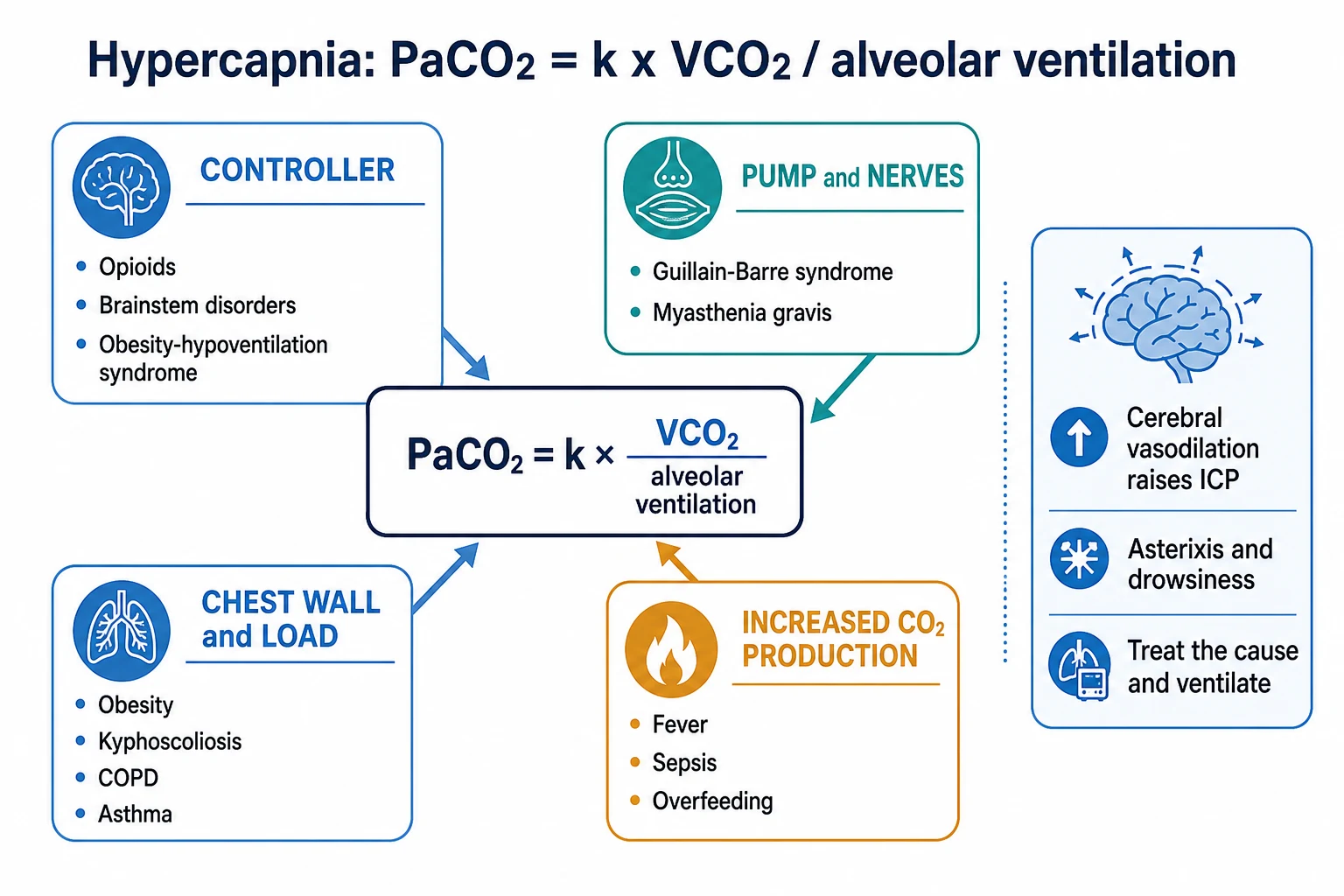
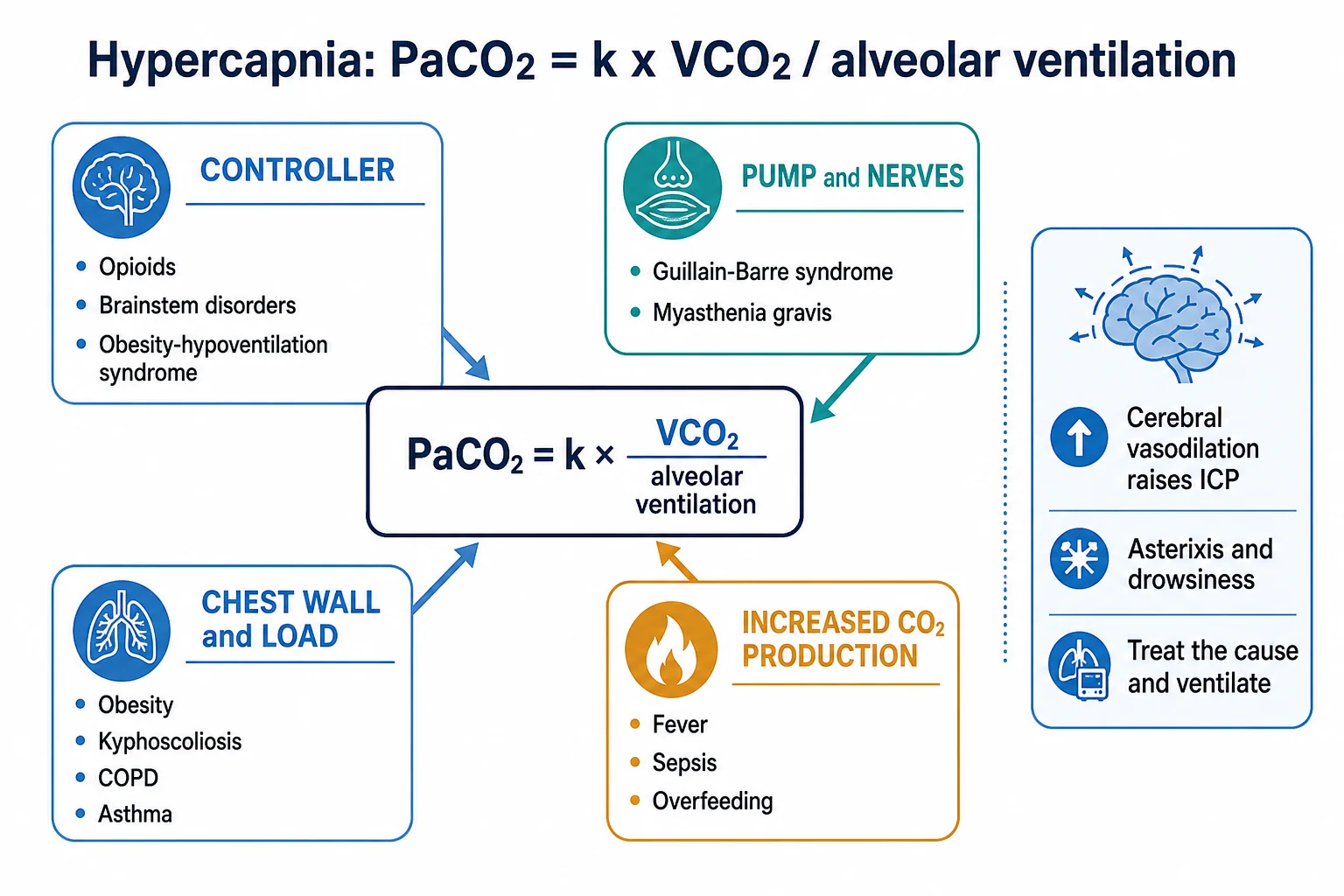
Hypercapnia is a raised arterial CO2 tension (PaCO2), and type-2 (ventilatory) respiratory failure is hypoxaemia with hypercapnia (a PaCO2 above about 45-50 mmHg, with a low pH). The governing equation is simple:[2][3]
PaCO2 is proportional to VCO2 divided by alveolar ventilation (PaCO2 = k x VCO2 / VA)[4]
So a high PaCO2 comes from either too much CO2 production or too little alveolar ventilation — the latter is by far the commoner, and it localises the failure to one of four sites: the controller, the pump and nerves, the chest wall and load, or the dead space.[2]
Causes — by the governing equation
Increased CO2 production (VCO2)
A high metabolic rate raises CO2 production: fever, sepsis, shivering, thyrotoxicosis, malignant hyperthermia, exercise, and overfeeding (excess carbohydrate raises the respiratory quotient and CO2 load). On its own this rarely causes hypercapnia, because the normal ventilatory response increases alveolar ventilation to clear the extra CO2 — it becomes a problem only when ventilation cannot rise (the critically ill or the fatiguing patient).[2]
Reduced alveolar ventilation — the commonest pathway
Alveolar ventilation falls at four sites, and the site determines whether the A-a gradient is normal or high:[1][2]
- The controller (central drive) — opioids, sedatives, brainstem stroke or lesion, central sleep apnoea, obesity-hypoventilation, hypothyroidism. The lungs are normal, so the A-a gradient is normal.
- The pump and nerves (neuromuscular) — Guillain-Barre syndrome, myasthenia gravis, motor neuron disease, critical-illness polyneuromyopathy, electrolyte disturbance (hypokalaemia, hypophosphataemia), residual muscle relaxants. The A-a gradient is normal.
- The chest wall and the load — obesity, kyphoscoliosis, ankylosing spondylitis, flail chest (the wall cannot expand); and increased airway resistance — COPD and asthma (the load of moving air through obstructed airways). Here the lung is diseased, so the A-a gradient is often high.
- Dead space — alveolar ventilation is (tidal volume minus dead space) times respiratory rate. If dead space rises (pulmonary embolism, emphysema, low cardiac output), effective alveolar ventilation falls even when minute ventilation is high. The A-a gradient is high.[4]
So: a pump or controller cause gives a normal A-a gradient; a lung, airway, or dead-space cause gives a high A-a gradient.[2]
Effects of hypercapnia
A rising PaCO2 has predictable physiological consequences:[2]
- Respiratory acidosis — a low pH (acute) with a normal or raised bicarbonate; the kidney retains bicarbonate over days to compensate (chronic).
- Cerebral vasodilation — CO2 is a potent cerebral vasodilator; hypercapnia raises cerebral blood flow and intracranial pressure (headache, papilloedema, confusion, and progressing to CO2 narcosis and coma).
- Sympathetic stimulation — tachycardia, sweating, hypertension, and arrhythmias (the early, easily-missed signs).
- Warm, flushed peripheries and a bounding pulse from vasodilation.
- Asterixis (a coarse flapping tremor) — a classic bedside sign of CO2 retention.
- Hypoxaemia — by the alveolar gas equation, a rising PaCO2 displaces the PAO2 down, so hypoxaemia accompanies hypercapnia.[3]
Clinical assessment
Suspect hypercapnia in the drowsy, flushed, asterixic patient with a bounding pulse, and confirm with an arterial blood gas (a raised PaCO2 with a low pH). Calculate the A-a gradient to localise the failure:[2]
- Normal A-a gradient — a controller or pump cause (opioids, neuromuscular weakness, obesity-hypoventilation).
- High A-a gradient — a lung, airway, or dead-space cause (COPD, asthma, pulmonary embolism).
Management
The principle is to ventilate the patient and reverse the cause.[2]
- Non-invasive ventilation (NIV) is the treatment of choice for hypercapnic COPD exacerbations — it reduces the work of breathing, corrects the pH, and reduces intubation and mortality.
- Invasive mechanical ventilation is needed if NIV fails, the patient is obtunded or unable to protect the airway, is aspirating, or is arresting.
- Treat the cause — naloxone for opioids, reversal of residual paralysis, treating sepsis and fever (to lower VCO2), antibiotics and bronchodilators for COPD, plasmapheresis for Guillain-Barre.
- Controlled oxygen in COPD — target an SpO2 of 88-92 per cent, to avoid oxygen-induced hypercapnia (below).
- Avoid bicarbonate — it is converted to CO2 and worsens hypercapnia; reserve it for the rare specific indication (for example, severe acidosis around intubation).[2]
- Reduce CO2 production — antipyretics, treat shivering and agitation, avoid carbohydrate overfeeding.
Oxygen-induced hypercapnia in COPD
Giving high-flow oxygen to a hypercapnic COPD patient can worsen the PaCO2 — the classic exam point. The mechanism is more than "loss of hypoxic drive":[2]
- The Haldane effect — oxygenated haemoglobin carries less CO2 than deoxygenated haemoglobin, so oxygenating the blood releases CO2 from haemoglobin into plasma, raising the PaCO2.
- Worsened V/Q mismatch — high oxygen relieves hypoxic pulmonary vasoconstriction, redistributing blood to poorly ventilated units and increasing dead space, which reduces effective alveolar ventilation.
- A reduced hypoxic drive — a smaller, contributory effect in some patients.
The practical point: give controlled oxygen (88-92 per cent SpO2) to the hypercapnic COPD patient, not high-flow oxygen.[2]
SAQ — The governing equation and localising the cause of hypercapnia
10 minutes · 10 marks
A 58-year-old man is brought to the emergency department drowsy and dyspnoeic. ABG on room air: pH 7.24, PaCO2 78 mmHg, PaO2 52, HCO3 28, BE +4. He takes morphine for chronic back pain. Examination reveals bilateral wheeze and a barrel chest. The examiners ask you to use the governing equation and the A–a gradient to localise the cause and to outline the differential diagnosis.
SAQ — Oxygen-induced hypercapnia and COPD exacerbation management
10 minutes · 10 marks
A 70-year-old woman with severe COPD is brought in drowsy (GCS 13) with an infective exacerbation. The ambulance crew gave high-flow oxygen; on arrival SpO2 is 98% on 15 L/min, with ABG pH 7.21, PaCO2 85, PaO2 110, HCO3 32. The examiners ask you to explain oxygen-induced hypercapnia, justify your oxygen target, and outline the role of non-invasive ventilation.
Red flags
Type-2 respiratory failure — formal definition and thresholds
Type-2 (ventilatory, hypercapnic) respiratory failure is defined by an arterial partial pressure of CO2 above 6.5 kPa (49 mmHg) that is not fully compensated — i.e. accompanied by a low or low-normal pH — with or without accompanying hypoxaemia (PaO2 <8 kPa / 60 mmHg).[10] The defining abnormality is alveolar hypoventilation, not a gas-exchange defect; the PaCO2 is the variable that has failed, and the pH tells you whether the failure is acute, chronic, or acute-on-chronic.
Type-2 respiratory failure: PaCO2 > 6.5 kPa (49 mmHg) with a low or low-normal pH (± hypoxaemia).[10]
The pH stratifies severity and guides the support in COPD exacerbations:[10]
- pH > 7.35 — PaCO2 elevated but compensated; standard medical therapy, no ventilatory support needed.
- pH 7.25-7.35 — the NIV window; bilevel NIV is first-line and reduces intubation and mortality.[7][8]
- pH < 7.25 — severe acidosis; NIV may still succeed in a monitored area, but a failing or obtunded patient needs intubation.
Type 1 — hypoxaemic
PaO2 <8 kPa, normal/low PaCO2
- Failure of oxygenation (V/Q mismatch, shunt, diffusion, low FiO2)
- High A-a gradient is the hallmark
- Causes: pneumonia, pulmonary oedema, ARDS, PE, pneumothorax, fibrosis
- Treat with oxygen, PEEP, treat the cause
Type 2 — hypercapnic
PaCO2 >6.5 kPa with low pH
- Failure of ventilation (alveolar hypoventilation)
- A-a gradient may be normal (controller/pump) or high (lung/airway/dead space)
- Causes: COPD, neuromuscular, opioids, obesity hypoventilation, brainstem
- Treat by ventilating (NIV/intubation) + reverse the cause; controlled O2 in COPD
Acute versus chronic respiratory acidosis — the compensation rules
The same PaCO2 means very different things depending on how quickly it rose. The kidney retains bicarbonate over two to three days to buffer the acid; so in acute hypercapnia the pH falls steeply, while in chronic hypercapnia (COPD, OHS) the raised bicarbonate keeps the pH near normal.[2][3]
Acute respiratory acidosis
metabolic compensation absent
- pH falls ~0.08 for every 10 mmHg (1.3 kPa) rise in PaCO2
- Bicarbonate rises only ~1 mEq/L per 10 mmHg (buffering, not renal)
- Example: PaCO2 70 mmHg → expected pH ~7.15, HCO3 ~26
- Clinical: opioid overdose, acute brainstem event, intubation mishap
Chronic respiratory acidosis
renal compensation complete
- pH falls only ~0.03 for every 10 mmHg rise in PaCO2 (often near-normal)
- Bicarbonate rises ~3.5-4 mEq/L per 10 mmHg (renal retention)
- Example: PaCO2 70 mmHg → expected pH ~7.36, HCO3 ~38
- Clinical: COPD, obesity hypoventilation, neuromuscular (MND), kyphoscoliosis
Acute-on-chronic
the common COPD scenario
- Raised baseline PaCO2 and HCO3 (compensated), now with a falling pH
- The drop in pH (not the absolute PaCO2) signals the acute decompensation
- Identify and treat the acute trigger: infection, pneumothorax, sedative, PE
- Correcting the PaCO2 to "normal" in a chronic retainer causes alkalaemia
The four big causes — worked clinical detail
COPD exacerbation — the prototypical type-2 failure
The commonest cause of acute hypercapnic respiratory failure on the ward and in the ED. An exacerbation narrows already-diseased airways (bronchoconstriction, inflammation, mucus), the work of breathing climbs, the respiratory muscles fatigue, and minute ventilation falls — the PaCO2 rises and the pH drops. Patients are chronic CO2 retainers with a raised baseline bicarbonate, so a "normal" PaCO2 of 50 in the recovering phase is acceptable; the danger is the falling pH.[2]
Key clues: known COPD, pursed-lip breathing, accessory-muscle use, pursed-lip breathing, prolonged expiration, a quiet chest (severe), wheeze, pedal oedema (cor pulmonale), and an SpO2 that has been pushed high by supplemental oxygen. Always ask what the trigger was — infection (most common), pneumothorax, pulmonary embolism, cardiac failure, sedative/opioid, or a missed inhaled therapy dose.[8]
Neuromuscular respiratory failure
The lung is normal; the pump fails. A-a gradient is normal. The signature is progressive loss of vital capacity with a rising PaCO2 as the breathing pattern fragments. Falling forced vital capacity (FVC), a maximal inspiratory pressure (MIP) below −30 cmH2O, or a rising PaCO2 are the triggers for ventilatory support — long before the patient looks distressed.[2][12]
Guillain-Barre (AIDP)
ascending weakness
- Monitor bedside FVC and MIP 4-hourly; intubate when FVC <15-20 mL/kg or MIP <-30
- Bulbar weakness and ineffective cough (MIC <40 L/min) predict aspiration
- Treat with IV immunoglobulin or plasma exchange; autonomic instability is common
Myasthenia gravis (myasthenic crisis)
fatigable weakness
- Ptosis, diplopia, dysarthria, fatigable limb weakness precede respiratory failure
- Negative inspiratory force and FVC trend the need for intubation
- Treat with pyridostigmine, IVIG/plasmapheresis, immunosuppression
Motor neuron disease / CIP/CIM
chronic or ICU-acquired
- MND: progressive respiratory-muscle weakness; consider NIV early for symptomatic hypoventilation
- Critical-illness polyneuromyopathy: weak limbs and poor weaning after prolonged ICU stay
- Exclude reversible metabolic causes (hypokalaemia, hypophosphataemia, hypomagnesaemia)
Drug-induced hypoventilation (opioids and sedatives)
A pure controller failure — the brainstem does not drive ventilation, so the patient is slow and shallow with a normal A-a gradient. Opioids (heroin, fentanyl, morphine, oxycodone), benzodiazepines, and residual neuromuscular blockade are the classic culprits. The picture is bradypnoea, miosis (opioids), reduced consciousness, and a PaCO2 that tracks the depth of sedation. Naloxone reverses opioids (titrate 40-100 mcg boluses to respiratory rate, not full alertness, to avoid precipitating acute withdrawal and pain); flumazenil reverses benzodiazepines (caution: seizures). Sugammadex or neostigmine reverses residual neuromuscular blockade. If reversal is incomplete or the airway is threatened, intubate rather than chase escalating naloxone doses.[2]
Obesity hypoventilation syndrome (OHS)
Defined as obesity (BMI >30) with daytime hypercapnia (PaCO2 >45 mmHg / 6 kPa) after excluding other causes of hypoventilation, usually with coexistent sleep-disordered breathing. The mechanism combines leptin resistance (loss of the ventilatory stimulant effect), mechanical load (chest-wall and abdominal mass raises the work of breathing), and blunted central drive. The Pickwick trial (Masa 2015) established that non-invasive ventilation (BiPAP) is superior to CPAP for most OHS patients, particularly those with severe nocturnal hypoventilation.[11][16]
The acute presentation mirrors COPD: hypercapnia, hypoxaemia, somnolence, cor pulmonale. BiPAP (IPAP 12-16, EPAP 6-8) unloads the load and restores drive; weight loss is the definitive therapy. Do not give uncontrolled high-flow oxygen — these patients have the same oxygen-induced hypercapnia risk as COPD.[11]
Permissive hypercapnia — when a high CO2 is the goal, not a failure
In severe ARDS, the lung-protective strategy (low tidal volume 6 mL/kg, low plateau pressure <30 cmH2O) deliberately underventilates to avoid volutrauma and barotrauma, and the PaCO2 is allowed to rise — this is permissive hypercapnia.[4][5] The principle: protecting the lung matters more than normalising the blood gas. Accept a PaCO2 of 60-80 mmHg (8-11 kPa) provided the pH stays above 7.20.[13][14]
The physiological case is stronger than just harm-avoidance: hypercapnic acidosis is itself protective — it attenuates stretch-induced and inflammatory lung injury, reduces reactive oxygen species, and down-regulates neutrophil function and cytokine release. Animal and observational data suggest a therapeutic effect, hence the framing "from permissive to therapeutic".[13][14]
Permissive hypercapnia (ARDS)
CO2 allowed to rise to protect the lung
- Low Vt 6 mL/kg IBW, plateau <30 cmH2O — the cause of the rising CO2
- Accept PaCO2 60-80 mmHg (8-11 kPa) if pH >7.20
- Hypercapnic acidosis is itself lung-protective and anti-inflammatory
- CONTRAINDICATED in raised intracranial pressure (cerebral vasodilation raises ICP)
Treat the hypercapnia (COPD, raised ICP)
CO2 must fall
- COPD exacerbation: NIV/intubation to correct the failing ventilation — the CO2 is the disease
- Raised ICP: normocapnia (PaCO2 35-40) prevents cerebral vasodilation and secondary brain injury
- Pulmonary hypertension/cor pulmonale: chronic hypercapnia drives remodelling
- Pregnancy: avoid maternal hypercapnia (fetal CO2 clearance depends on the gradient)
The single absolute contraindication to permissive hypercapnia is raised intracranial pressure — every 1 mmHg rise in PaCO2 increases cerebral blood flow by ~2-4 per cent and raises ICP. In traumatic brain injury, subarachnoid haemorrhage, and hepatic encephalopathy with cerebral oedema, keep PaCO2 35-40 mmHg.[17]
Non-invasive ventilation (BiPAP) — IPAP and EPAP demystified
In acute hypercapnic respiratory failure, NIV is bilevel (BiPAP), not CPAP. Two pressures alternate with the patient's breath:[10][12]
- IPAP (inspiratory positive airway pressure) — the pressure delivered on inspiration. It unloads the fatigued inspiratory muscles and boosts tidal volume, which is what lowers the PaCO2. Titrate IPAP up to drop the CO2.
- EPAP (expiratory positive airway pressure) — the baseline (PEEP-like) pressure. It splints the airways open, recruits underventilated alveoli, and counteracts intrinsic (auto-) PEEP in COPD, lowering the threshold the patient must generate to trigger a breath. Raise EPAP to improve oxygenation.
IPAP lowers the CO2; EPAP raises the oxygen. In COPD, EPAP (4-6) is set just below the intrinsic PEEP to "pop" the obstructed airways without adding gas trapping.[12]
BiPAP starting settings in acute type-2 failure
Starting and titrating bilevel NIV in acute hypercapnic failure
Step 1 — Set up the patient and the mask
Sit the patient upright. Choose a well-fitting oronasal (full-face) mask first. Let the patient hold the mask to their face to acclimatise before strapping it on. Explain what the machine does — anxiety and claustrophobia are the commonest early failures.
Step 2 — Choose initial pressures
COPD/OHS: IPAP 10-12, EPAP 4-6 cmH2O. Neuromuscular/chest-wall: IPAP 10-12, EPAP 4 (less intrinsic PEEP). Backup rate 12-14, FiO2 to SpO2 88-92 per cent (COPD) or 94-98 per cent (others). Ramp/timing as tolerated.
Step 3 — Titrate IPAP to drop the CO2
Increase IPAP by 2 cmH2O every 10-15 minutes (toward 20) until the tidal volume rises, the respiratory rate falls toward <25, and the patient looks less distressed. Keep EPAP at 4-6 in COPD; raise EPAP only if oxygenation is the problem.
Step 4 — Reassess at one hour
Repeat the ABG and examine the patient. Improved (rising pH, falling PaCO2, falling RR, less distress) — continue and plan to wean. Not improving (pH still <7.25, rising PaCO2/RR, falling GCS) — this is NIV failure: escalate to intubation, do not prolong a failing trial. The pH at one hour is the best predictor of success.
Step 5 — Wean as the cause reverses
Once pH >7.35 and PaCO2 trending down, reduce IPAP in 2 cmH2O steps, lengthen off-periods, and use a standard mask for meals. Treat the trigger (antibiotics, bronchodilators, steroids, naloxone). Do not stop NIV abruptly in a chronic retainer.
Contraindications to NIV
Absolute (intubate instead): respiratory arrest or peri-arrest; inability to protect the airway (GCS <8, copious secretions, vomiting); severe agitation or confusion precluding cooperation; haemodynamic instability; facial trauma/surgery or fixed upper-airway obstruction.[10]
Relative (proceed cautiously in a monitored area): pH <7.25, radiographic pneumonia, the patient who fails to improve at one hour, recent upper-GI surgery.[15]
Recognising and managing NIV failure
NIV fails in 15-25 per cent of acute hypercapnic COPD. The skill is recognising failure early — a delayed intubation after a failed NIV trial is a higher-risk, worse-outcome intubation than a timely one.[7][8]
Intubate when any of:
- pH <7.25 and not improving after one hour of optimised NIV — the single best objective criterion.
- Worsening consciousness / falling GCS — CO2 narcosis; the airway is no longer protected.
- Respiratory arrest or peri-arrest — bradypnoea, silent chest, loss of effort.
- Haemodynamic instability — hypotension, sustained tachycardia, new arrhythmia.
- Copious secretions NIV cannot clear, or agitation/confusion precluding mask tolerance.[8]
Complications of hypercapnia — the end-organ effects
Hypercapnia is not benign. A sustained high PaCO2 damages the lung circulation, the heart, and the brain.[14]
Pulmonary vascular — vasoconstriction, pulmonary hypertension, cor pulmonale
Hypercapnia (with the associated acidosis) is a pulmonary vasoconstrictor — the mirror image of its systemic vasodilator effect. Chronic hypercapnia (COPD, OHS) drives remodelling of the pulmonary vascular bed, raising pulmonary vascular resistance and producing pulmonary hypertension and right-heart failure (cor pulmonale). The clinical signature is a raised JVP, a parasternal heave, a loud P2, tricuspid regurgitation, and peripheral oedema. Reversing the hypercapnia (NIV, treating the cause) is part of the treatment.[2]
Cardiac — arrhythmias and sympathetic surges
Hypercapnia and its acidosis increase myocardial excitability: atrial and ventricular ectopics, atrial fibrillation, and — at extreme PaCO2 — ventricular tachycardia. The early sympathetic surge (tachycardia, hypertension, sweating) gives way to myocardial depression and vasodilation as the acidosis deepens. Hyperkalaemia (acidosis-driven) and catecholamine surges combine to lower the threshold for arrhythmia. Correct the CO2 and the pH, not the rhythm in isolation.[2]
Cerebral — intracranial hypertension and CO2 narcosis
CO2 is the dominant physiological regulator of cerebral vascular tone: every 1 mmHg rise in PaCO2 increases cerebral blood flow by ~2-4 per cent, raising intracranial volume and pressure. Acute hypercapnia produces headache, confusion, and asterixis (the coarse flapping tremor of CO2 retention), progressing through somnolence and CO2 narcosis to coma. In a patient with an intracranial mass or brain injury, even modest hypercapnia can precipitate herniation. Papilloedema is a late sign of chronic hypercapnia (a cause of "idiopathic" intracranial hypertension in OHS).[2][17]
ABG interpretation in type-2 failure — a worked method
The arterial blood gas in suspected type-2 failure answers four questions in sequence:[2][3]
- Is the PaCO2 high? (>45 mmHg / 6 kPa) — confirms ventilatory failure.
- What is the pH? — low (acidosis) means the failure is uncompensated/acute or acute-on-chronic; near-normal means chronic and compensated.
- What is the bicarbonate? — raised means renal compensation is present (chronic); normal means acute.
- What is the A-a gradient? — normal points to controller/pump (opioids, neuromuscular, OHS, brainstem); high points to lung/airway/dead-space (COPD, asthma, pneumonia, PE).
Expected compensation (to detect a mixed disorder): in acute respiratory acidosis, pH falls ~0.08 and HCO3 rises ~1 mEq/L per 10 mmHg rise in PaCO2; in chronic respiratory acidosis, pH falls ~0.03 and HCO3 rises ~3.5-4 mEq/L per 10 mmHg. A pH or HCO3 outside these bands means a mixed disorder (e.g. a chronic retainer with a superimposed metabolic acidosis from sepsis or renal failure).[12]
Evidence and landmark trials
Brochard 1995
NEJM 1995
85 pts with AECOPD randomised to face-mask pressure-support NIV vs standard therapy
Key finding
NIV reduced complications, shortened hospital stay, and showed a trend to lower mortality; fewer required intubation
Practice change
Established NIV as superior to standard therapy in moderate-severe AECOPD
PLANT (Plant 2000)
Lancet 2000
236 pts, pH 7.25-7.35 AECOPD on general respiratory wards — multicentre RCT of early NIV vs standard therapy
Key finding
Intubation 15% NIV vs 27% control (p=0.05); in-hospital mortality 4.7% lower; fewer complications; shorter stay
Practice change
Made early bilevel NIV first-line for the acidotic AECOPD on the ward
Lightowler (Cochrane/BMJ 2003)
BMJ 2003
Meta-analysis of 8 RCTs — NPPV + usual care vs usual care for hypercapnic AECOPD
Key finding
Reduced mortality (RR 0.41), intubation (RR 0.42), treatment failure (RR 0.51), and length of stay (-3.24 days); rapid pH/PaCO2/RR improvement at 1 hour
Practice change
Confirmed NIV + usual care as first-line for all suitable AECOPD patients
Austin 2010
BMJ 2010
405 pts with presumed COPD — prehospital titrated oxygen (target SpO2 88-92%) vs high-flow oxygen RCT
Key finding
Mortality 9% (titrated) vs 12% (high-flow); adjusted OR 0.42 for death — high-flow oxygen was harmful
Practice change
Reinforced controlled oxygen (88-92%) in suspected COPD from first contact, including the ambulance
Amato 1998
NEJM 1998
53 pts with early ARDS — protective ventilation (low Vt, PEEP above LIP, permissive hypercapnia) vs conventional ventilation
Key finding
28-day mortality 38% protective vs 71% conventional (p=0.005); barotrauma and weaning favouring protective strategy
Practice change
Introduced the lung-protective, permissive-hypercapnia paradigm for ARDS
ARDSNet ARMA 2000
NEJM 2000
861 pts with ALI/ARDS — Vt 6 vs 12 mL/kg PBW, plateau <30 vs <50 cmH2O
Key finding
Mortality 31% low-Vt vs 40% traditional (p=0.007); more ventilator-free days
Practice change
Made 6 mL/kg PBW low-tidal-volume ventilation the standard of care for ARDS (with attendant permissive hypercapnia)
Confalonieri 1999
AJRCCM 1999
56 pts with severe CAP and respiratory failure — NIV vs standard therapy RCT
Key finding
Reduced intubation (21% vs 50%), ICU stay, and 1-year mortality; benefit greatest in COPD + CAP
Practice change
Extended the NIV indication to selected hypercapnic severe-CAP patients, especially with underlying COPD
Pickwick (Masa 2015)
AJRCCM 2015
221 OHS pts (PaCO2 >45, AHI >30) — NIV vs CPAP for 2 months, multicentre RCT
Key finding
Greater improvement in PaCO2 and bicarbonate with NIV; NIV preferable in severe nocturnal hypoventilation, CPAP acceptable if hypercapnia resolves on CPAP
Practice change
Established BiPAP as the preferred initial ventilatory mode for OHS with significant hypoventilation
Pitfalls and exam favourites
[8]Clinical pearls
References
- [1]Petersson J, Glenny RW. Gas Exchange in the Lung. Seminars in Respiratory and Critical Care Medicine, 2023.PMID 37816345
- [2]Karnad DR, Nor MBM, Richards GA, et al. Intensive care in severe malaria: Report from the task force on tropical diseases by the World Federation of Societies of Intensive and Critical Care Medicine. Journal of critical care, 2018.PMID 29132978
- [3]Bigatello L, Pesenti A Respiratory Physiology for the Anesthesiologist. Anesthesiology, 2019.PMID 30998510
- [4]Brower RG, et al. (Acute Respiratory Distress Syndrome Network) Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. New England Journal of Medicine, 2000.PMID 10793162
- [5]Amato MB, Barbas CS, Medeiros DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. New England Journal of Medicine, 1998.PMID 9449727
- [6]Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. New England Journal of Medicine, 1995.PMID 7651472
- [7]Plant PK, Owen JL, Elliott MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet, 2000.PMID 10859037
- [8]Lightowler JV, Wedzicha JA, Elliott MW, Ram FS. Non-invasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ, 2003.PMID 12543832
- [9]Austin MA, Wills KE, Blizzard L, Walters EH, Wood-Baker R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: randomised controlled trial. BMJ, 2010.PMID 20959284
- [10]Davidson AC, Banham S, Elliott M, et al. BTS/ICS guideline for the ventilatory management of acute hypercapnic respiratory failure in adults. Thorax, 2016.PMID 26976648
- [11]Mokhlesi B, Masa JF, Brozek JL, et al. Evaluation and management of obesity hypoventilation syndrome. An official American Thoracic Society clinical practice guideline. American Journal of Respiratory and Critical Care Medicine, 2019.PMID 31368798
- [12]Nava S, Hill N. Non-invasive ventilation in acute respiratory failure. Lancet, 2009.PMID 19616722
- [13]Ijland MM, Lachmann B, Herting E, van der Hoeven JG. Bench-to-bedside review: hypercapnic acidosis in lung injury - from 'permissive' to 'therapeutic'. Critical Care, 2010.PMID 21067531
- [14]Laffey JG, O'Croinin D, McLoughlin P, Kavanagh BP. Permissive hypercapnia - role in protective lung ventilatory strategies. Intensive Care Medicine, 2004.PMID 14722644
- [15]Confalonieri M, Potena A, Carbone G, et al. Acute respiratory failure in patients with severe community-acquired pneumonia. A prospective randomized evaluation of noninvasive ventilation. American Journal of Respiratory and Critical Care Medicine, 1999.PMID 10556125
- [16]Masa JF, Corral J, Alonso ML, et al. Efficacy of different treatment alternatives for obesity hypoventilation syndrome. The Pickwick study. American Journal of Respiratory and Critical Care Medicine, 2015.PMID 25915102
- [17]Go SL, Harris DS, Bruder N, Singh JM. Pro/con debate: should PaCO2 be tightly controlled in all patients with acute brain injuries? Critical Care, 2013.PMID 23360555