Paeds · allergy-and-immunology
Antibody deficiencies
Also known as Humoral immunodeficiency · Primary antibody deficiency · Hypogammaglobulinaemia · Agammaglobulinaemia · Inborn errors of humoral immunity
A fellowship approach to antibody deficiencies in children: recognise the recurrent-infection pattern that warrants an immunoglobulin work-up, quantify the defect with total immunoglobulins plus functional vaccine response rather than treating a single low number, classify into a primary inborn error (XLA, CVID, specific antibody deficiency, IgA deficiency, transient hypogammaglobulinaemia of infancy, hyper-IgM) or a secondary cause, then escalate only the children who genuinely need immunoglobulin replacement while protecting the lung and planning transition.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A two-year-old boy is on his fourth course of antibiotics this year for otitis media and has just had his second pneumonia. His immunoglobulins come back with a total IgG well below the age-adjusted reference range. Two doors open from that single result. Through one door sits a child with X-linked agammaglobulinaemia who will need lifelong immunoglobulin replacement and genetic counselling for his mother's carrier relatives. Through the other sits a thriving toddler with transient hypogammaglobulinaemia of infancy who will be normal by age four and needs only a watchful plan. Telling those two children apart — without over-investigating one and under-treating the other — is the whole skill of this topic. [1] [2]
A.N.T.I.B.O.D.Y.
Overview & Definition
An antibody deficiency is a state in which the body cannot produce adequate functional antibody to clear the organisms that antibody normally handles — chiefly encapsulated bacteria such as Streptococcus pneumoniae and Haemophilus influenzae, and enteric viruses. The defect may sit anywhere along B-cell development, from the stem-cell stage through to the class-switched plasma cell. Wherever it sits, the clinical fingerprint is the same: recurrent, persistent, severe, or unusual infection, most often of the sinopulmonary tract. [2]
The headline distinction for a general paediatrician is between quantitative defects (the immunoglobulin levels are low) and functional defects (the levels are normal but the antibody does not work). Specific antibody deficiency is the classic functional defect — a child with normal total IgG who nevertheless fails to respond to polysaccharide antigens. This is why a functional vaccine challenge belongs in every work-up, not only when the numbers look low. [4] [10]
Primary antibody deficiencies sit within the International Union of Immunological Societies (IUIS) classification of inborn errors of immunity, most of them in the category of predominantly antibody deficiencies. The 2022 update lists the agreed clinical and laboratory phenotypes, and it is this framework — not a single test — that a fellowship answer is built on. [2]
Classification
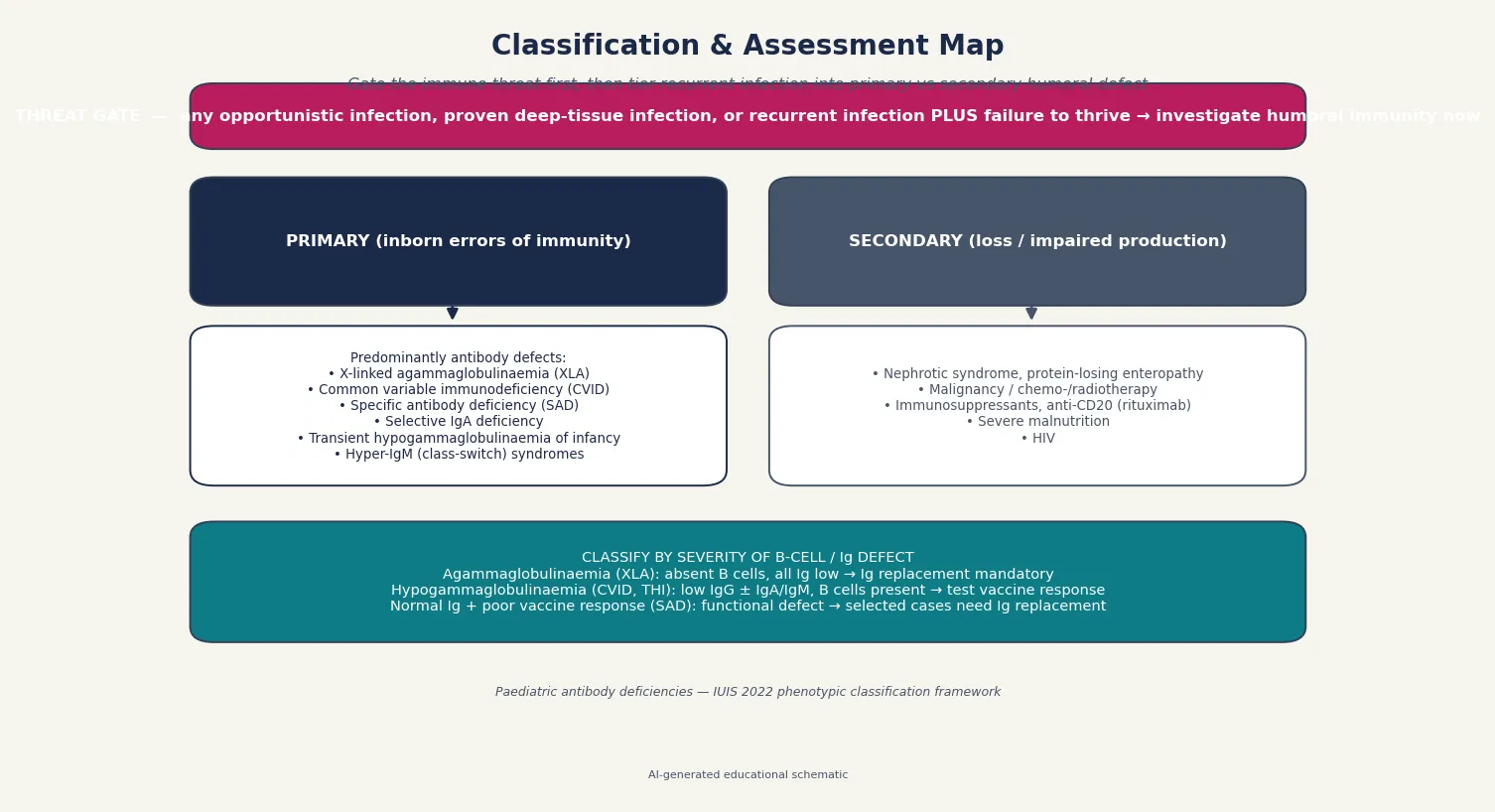
Sort antibody deficiency by the site of the B-cell block, because that single axis predicts the immunoglobulin pattern, the inheritance, and the urgency of replacement. The 2022 IUIS classification organises predominantly antibody deficiencies into a small number of recognisable phenotypes. [2]
Agammaglobulinaemia describes the near-total absence of immunoglobulin and B cells. The prototypic cause is X-linked agammaglobulinaemia (XLA), caused by a block in the BTK gene that arrests B-cell development at the pro-B to pre-B stage. Affected males present after maternal antibody wanes, around four to twelve months, with deep-tissue infection and characteristically absent tonsils and lymph nodes. Autosomal recessive agammaglobulinaemia is rarer and phenotypically similar. [3]
Hypogammaglobulinaemia with B cells present is the larger and messier group. Common variable immunodeficiency (CVID) is the commonest symptomatic primary antibody deficiency requiring treatment: low IgG with low IgA and/or IgM, impaired vaccine response, and a onset that is typically later, in the second decade or beyond, although childhood presentations occur. Transient hypogammaglobulinaemia of infancy (THI) is a self-limiting maturational delay in which IgG is low but B cells are present and vaccine response is preserved; it resolves by age three to four years and needs no replacement. [6] [5]
Selective IgA deficiency is the commonest primary antibody defect overall, defined by an isolated low IgA with normal IgG and IgM. Most affected children are asymptomatic; some have recurrent mucosal infection or coexist with allergy and autoimmune disease. Specific antibody deficiency (SAD) is the functional counterpart: normal immunoglobulin quantities with a documented failure to mount antibody against polysaccharide antigens. The hyper-IgM syndromes, such as CD40 ligand deficiency, fail class-switch recombination and produce low IgG and IgA with normal or frankly elevated IgM. [2] [10]
Finally, separate secondary antibody loss — nephrotic syndrome, protein-losing enteropathy, malignancy and its treatment, immunosuppressive drugs including rituximab, severe malnutrition, and HIV — from the primary inborn errors. The distinction matters because the secondary group often recovers when the underlying cause is treated, and because immunoglobulin replacement is rarely the long-term answer for them. [6]
Epidemiology & Risk Factors
Primary antibody deficiencies are individually rare but collectively common enough that every general paediatrician will meet several in a career. Boyle and Buckley, using United States registry data, estimated the diagnosed prevalence of primary immunodeficiency diseases and showed that antibody defects dominate the overall burden. Most cases are not diagnosed at first presentation, which is the central reason the warning signs exist. [1]
Selective IgA deficiency is the commonest of all, with a prevalence around one in five hundred to one in six hundred in European-ancestry populations, and most affected individuals never come to medical attention. CVID is the commonest symptomatic primary antibody deficiency that requires treatment, with an estimated prevalence of around one in twenty-five thousand. XLA, by contrast, is rare, with an incidence of roughly one in two hundred thousand live male births, but it is the one you cannot afford to miss. [3] [2]
Risk factors are family history and consanguinity. A family history of early male infant death, a known primary immunodeficiency, or parental consanguinity should lower the threshold for quantifying immunoglobulins in a child with recurrent infection. The age of the child matters too: physiologically low IgG is normal in infants between three and six months as maternal antibody falls, and a single low value in that window is expected rather than pathological. [1] [9]
The two warning-sign frameworks — the Jeffrey Modell Foundation ten warning signs and the modified Düsseldorf criteria — were built to turn recurrent infection into a decision to investigate. The Düsseldorf group showed that the original ten signs had only modest sensitivity, with failure to thrive and a positive family history performing best, and that recurrent infection in an otherwise well child still warrants a basic immunoglobulin screen. The message is to use the signs as a prompt, not a gatekeeper. [8] [9]
Pathophysiology
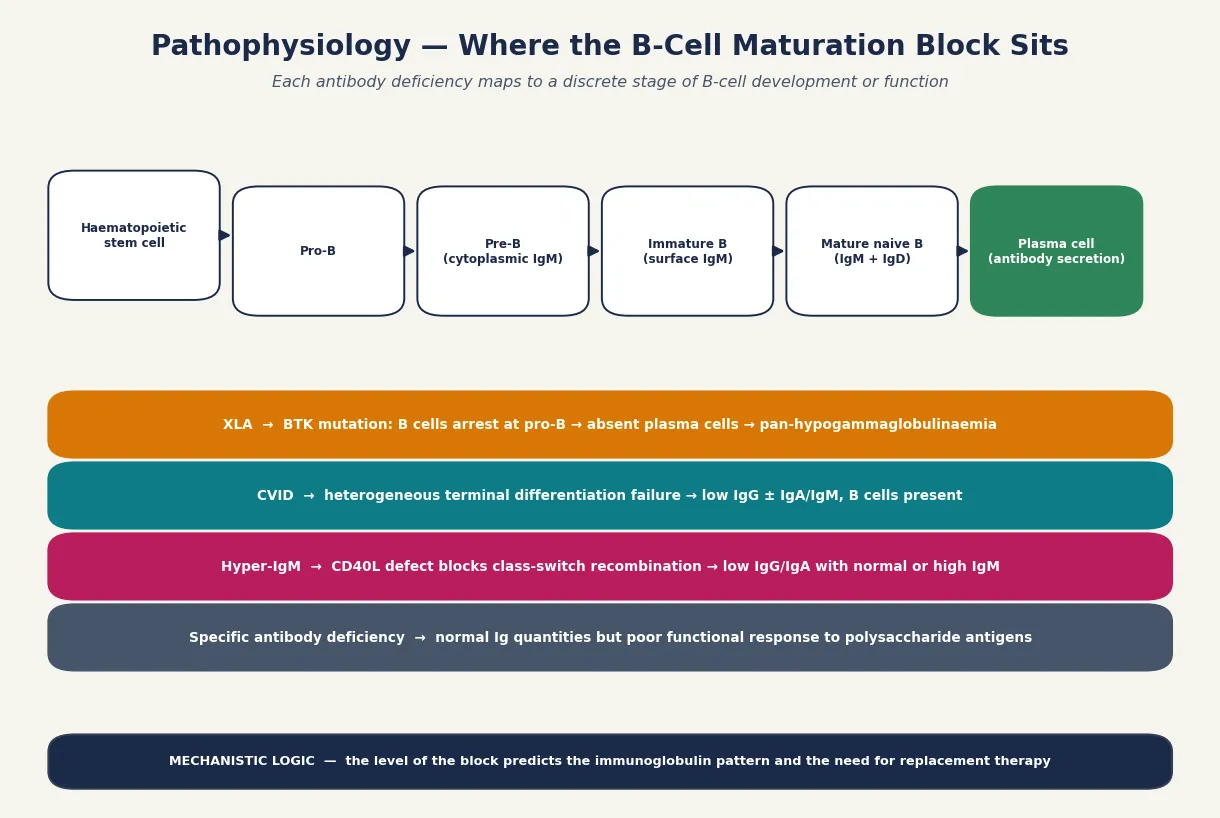
Antibody deficiencies arise from blocks along B-cell development, and the site of the block dictates the immunoglobulin pattern you measure at the bench. A fellowship answer earns its marks by linking the developmental stage to the laboratory phenotype. [2]
In X-linked agammaglobulinaemia, a mutation in the BTK gene stops B cells completing the transition from pro-B to pre-B cell in the bone marrow. Because the lineage never matures, no B cells ever reach the periphery, no plasma cells form, and immunoglobulin of all classes is near-absent. The clinical consequence is severe, recurrent, deep-tissue infection beginning once transplacentally acquired maternal IgG clears, typically between four and twelve months of age. The absent tonsils and palpable lymph nodes are the physical sign of a B-cell compartment that was never built. [3]
Common variable immunodeficiency is mechanistically heterogeneous. The terminal differentiation of B cells into antibody-secreting plasma cells and class-switched memory cells fails, even though B-cell numbers are usually normal or only mildly reduced. The result is low IgG with low IgA and/or IgM, and a poor functional response to both protein and polysaccharide vaccines. Unlike XLA, the onset is later — rarely before the second year and often in adolescence or adulthood — because the early B-cell machinery is intact. [4] [5]
The hyper-IgM syndromes illustrate class-switch failure. In CD40 ligand deficiency, T cells cannot deliver the signal that drives B cells to switch from IgM production to IgG and IgA. Immunoglobulin G and A are therefore low while IgM is normal or elevated, and because CD40 signalling also governs macrophage and T-cell responses to intracellular organisms, these children are additionally vulnerable to Pneumocystis and cryptosporidium. Specific antibody deficiency, by contrast, is a pure functional defect: immunoglobulin is made in normal quantities but the response to polysaccharide antigens is impaired, leaving the child unable to clear encapsulated bacteria despite a reassuring-looking IgG. [2] [10]
Transient hypogammaglobulinaemia of infancy is a maturational delay rather than a fixed defect. B-cell development is intact but endogenous IgG production lags, so the infant passes through a nadir of low IgG with preserved function before catching up by age three to four. Recognising this self-limiting course is what prevents an unnecessary commitment to lifelong immunoglobulin therapy. [6]
Clinical Presentation
The clinical fingerprint of an antibody deficiency is recurrent, persistent, severe, or unusual infection, and the most reliable place to find that pattern is the sinopulmonary tract. Otitis media, sinusitis, and pneumonia recur, persist beyond the expected course, and leave structural damage such as bronchiectasis if the defect is not addressed. [3] [7]
X-linked agammaglobulinaemia declares itself after maternal antibody wanes. A previously well boy develops deep-tissue infection — pneumonia, sepsis, meningitis, septic arthritis, or osteomyelitis — between four and twelve months, often with organisms such as Pneumococcus, Haemophilus, and Staphylococcus. The examination is distinctive: tonsils and lymph nodes are small or absent, and chronic enteroviral infection can produce a chronic meningoencephalitis that is characteristic of the condition. [3]
Common variable immunodeficiency presents later and more variably. Recurrent sinopulmonary infection is the core feature, but CVID also carries a substantial burden of autoimmune disease (immune thrombocytopenia, autoimmune haemolytic anaemia), granulomatous and lymphoproliferative disease, and an increased risk of lymphoma. A teenager with recurrent pneumonia plus autoimmune cytopenia should prompt immunoglobulin testing. [4] [5]
Selective IgA deficiency is usually silent. Symptomatic children tend to have recurrent mucosal infection, an association with allergy and autoimmune disease, and — of practical importance — a risk of anti-IgA antibodies that can cause anaphylaxis on blood product transfusion. Specific antibody deficiency mirrors recurrent sinopulmonary and gastrointestinal infection despite normal immunoglobulin levels. The hyper-IgM syndromes combine recurrent bacterial infection with opportunistic infection such as Pneumocystis pneumonia and neutropenia. [2] [10]
Transient hypogammaglobulinaemia of infancy sits at the benign end. The infant has recurrent or persistent minor infection during the physiological IgG nadir, but thrives, responds normally to vaccines, and normalises by age three to four. The risk is not the condition itself but the harm of labelling it as permanent and starting immunoglobulin unnecessarily. [6]
Differential Diagnosis
The differential for recurrent infection in a child is broad, and the discipline of this topic is to think structurally rather than to chase antibody deficiency too early. Normal childhood exposure, anatomical causes, atopy, and secondary immunodeficiency all produce recurrent symptoms and most of them outnumber the primary antibody defects. [8]
Physiological recurrent infection is the commonest explanation. A child in daycare or with school-age siblings can have eight to ten viral upper-respiratory infections a year, each lasting up to two weeks, and still be normal. The discriminating features are severity (deep-tissue infection), persistence (infection that fails to clear), and the organism (encapsulated bacteria, opportunists, or normally vaccine-preventable organisms). [9]
Anatomical and barrier causes mimic immunodeficiency. Recurrent pneumonia in the same lobe raises the question of an inhaled foreign body, a congenital lung malformation, or a airway anomaly. Recurrent skin infection may reflect eczema as a portal of entry, and chronic suppurative ear disease may reflect eustachian-tube dysfunction or a structural problem. These are excluded by history, examination, and targeted imaging. [6]
Atopy is a frequent confounder. Allergic rhinitis produces the congested, sniffly, chronically blocked child whose "constant cold" is actually allergic inflammation, and who improves with anti-inflammatory treatment rather than antibiotics. A careful allergy history and examination usually separate this from true infection susceptibility. [1]
Secondary immunodeficiency must be excluded before a primary defect is confirmed. Nephrotic syndrome and protein-losing enteropathy waste immunoglobulin; malignancy, chemotherapy, and radiotherapy suppress production; rituximab depletes B cells; severe malnutrition impairs immunity globally; and HIV produces a combined defect. A primary antibody deficiency should not be diagnosed until these are considered, because the treatment of the secondary cause is usually more important than immunoglobulin replacement. [6] [10]
| Pattern | Likely alternative | Key discriminator |
|---|---|---|
| 8-10 viral colds/year, daycare | Normal childhood exposure | Well between episodes, full recovery, normal growth |
| Same-lobe pneumonia repeatedly | Inhaled foreign body or malformation | Localised finding, inspiratory trap on imaging |
| Generalised oedema + recurrent infection | Nephrotic syndrome (secondary loss) | Proteinuria, hypoalbuminaemia, low IgG |
Clinical & Bedside Assessment
The bedside task is to decide whether the recurrent-infection pattern crosses the threshold for an immunoglobulin work-up, and to gather the discriminating evidence at the first consultation. Begin with the threat gate: any proven deep-tissue infection, opportunistic organism, or infection with a normally vaccine-preventable organism earns a work-up regardless of how well the child looks between episodes. [3] [8]
The history is the discriminating instrument. Establish the number, site, severity, and documented microbiology of infections, and whether each required oral or intravenous antibiotics or admission. Ask specifically about pneumonia confirmed on imaging, deep-tissue infection, and chronic diarrhoea. Take a family history for early male death, known primary immunodeficiency, and consanguinity, and a developmental and growth history, because failure to thrive alongside infection changes the threshold to investigate immediately. [9]
The examination carries the physical clues that point to specific entities. Assess tonsil and lymph-node size — their absence in a boy with recurrent deep infection is the classic sign of XLA. Look for the sequelae of chronic infection: clubbing, chest wall deformity, localised crackles, and signs of bronchiectasis. Examine growth parameters and plot them, because faltering growth with infection is a red flag. Look for autoimmune features such as bruising or pallor that broaden the picture toward CVID. [3] [4]
Assess the family's practical context at the same visit. A child in a remote community, a refugee family, or a household with socioeconomic disadvantage may present late and face barriers to long-term immunoglobulin access. Documenting these factors at the first assessment shapes the disposition, the retrieval plan, and the transition to shared care. [6]
The decision to investigate rests on combining the warning signs with clinical judgement. The ten Jeffrey Modell warning signs and the modified Düsseldorf criteria provide a prompt, but the strongest single signals are failure to thrive, a positive family history, and infection with an unusual organism. Use the signs to lower the threshold for a simple, cheap immunoglobulin screen rather than as an absolute checklist. [8] [9]
Investigations
The investigation ladder is designed to answer a single functional question first: can this child make antibody? Begin with quantification, add function, and only then move to cellular and genetic characterisation. [6]
First-line tests are a full blood count with differential, total immunoglobulins (IgG, IgA, IgM) interpreted against age-adjusted reference ranges, and a functional vaccine response. The vaccine response is the decisive test. Measure specific IgG to Streptococcus pneumoniae before and two to four weeks after a dose of pneumococcal polysaccharide vaccine (for children over two years who have completed conjugate priming); a failure to respond to the majority of serotypes defines a functional defect even when total IgG is normal. [4] [10]
Second-line tests characterise the cellular defect. Lymphocyte subset enumeration by flow cytometry counts B cells (CD19, CD20), T cells, and natural killer cells. A profoundly low or absent B-cell count separates agammaglobulinaemia (XLA and its autosomal recessive mimics) from hypogammaglobulinaemia with B cells present (CVID, THI). Further subclass testing and memory-B-cell phenotyping refine the picture in CVID and specific antibody deficiency. [2] [6]
Genetic testing confirms monogenic causes and enables family counselling and cascade screening. XLA is confirmed by identifying a pathogenic BTK variant, and hyper-IgM syndromes by variants in CD40L and related genes. Next-generation sequencing gene panels are now the standard approach when a primary antibody deficiency is suspected, and they distinguish the monogenic agammaglobulinaemias from genetically unresolved CVID. [2]
Imaging and lung surveillance are investigations of complication and severity, not diagnosis. A baseline chest radiograph, spirometry in cooperative children, and low-dose chest computed tomography when bronchiectasis is suspected document structural lung disease and guide the intensity of therapy. The European Chest CT Group data show that bronchial pathology is common and often under-recognised in antibody-deficient patients, which is why imaging is part of the work-up rather than an afterthought. [7]
Management — Resuscitation
Resuscitation in antibody deficiency means treating the acute presentation safely while avoiding the two harms that are specific to this group: anaphylaxis on blood products in IgA-deficient patients, and live vaccines in a child with an undiagnosed combined defect. [2] [5]
A child presenting with acute severe infection receives the standard paediatric sepsis pathway: prompt cultures, empirical intravenous antibiotics, and fluid and oxygen as required. The microbiology is guided by the likely organisms — encapsulated bacteria in pure antibody deficiency, opportunists in combined defects. Empirical cover should be broadened when an opportunistic organism is suspected, and intravenous immunoglobulin may be added for acute severe infection in a known antibody-deficient child, although this is distinct from the maintenance replacement therapy discussed below. [3]
The antibody-specific resuscitation hazard is transfusion in selective IgA deficiency. Some IgA-deficient patients develop anti-IgA antibodies that can trigger anaphylaxis on exposure to IgA-containing blood products. If a patient with known IgA deficiency needs transfusion, use washed red cells, IgA-depleted products, or autologous blood, and ensure the team is aware of the risk. [2]
The second hazard is live vaccination in a possible combined immunodeficiency. If there is any suspicion of a combined T- and B-cell defect — lymphopenia, failure to thrive, opportunistic infection — defer live vaccines such as rotavirus, measles-mumps-rubella, and BCG until the immune status is clarified, because live vaccines can cause disseminated disease in severely immunocompromised infants. [2] [6]
Resuscitation of the family's understanding matters too. A child newly diagnosed with an antibody deficiency will need a structured plan, and parents benefit from an early, honest explanation of what the diagnosis means, what the treatment involves, and what the prognosis is, before the detail of long-term management is introduced. [5]
Management — Definitive & Stepwise
Definitive management follows a five-step ladder that a fellowship candidate can recite: confirm the defect, replace antibody where indicated, suppress infection, protect the lung, and plan the transition. The discipline is to escalate only the children who need it. [5] [6]
Step one is to confirm the defect before treating it. Immunoglobulin replacement is a lifelong, resource-intensive therapy with real risks, and it should not start on the basis of a single low number. Confirm the defect with quantified immunoglobulins plus a functional vaccine response, exclude secondary causes, and characterise the cellular and genetic defect where possible. Only a child with demonstrated infection susceptibility and a confirmed quantified defect moves to replacement. [4] [10]
Step two is immunoglobulin replacement for the child who needs it. The principal indications are XLA and other agammaglobulinaemias, and CVID with documented infection susceptibility. Replacement is given intravenously (IVIG) or subcutaneously (SCIG), dosed to keep the trough IgG within the normal range and individualised to the child's infection pattern and pharmacokinetics. Typical IVIG dosing is in the range of 400 to 600 mg/kg every three to four weeks, with SCIG given weekly at an equivalent monthly dose, though the exact regimen is tailored by trough levels and clinical response. [3] [5]
Step three is infection suppression and prophylaxis. Vaccinate household contacts to provide herd protection, because the antibody-deficient child may not respond reliably to vaccines themselves (and must not receive live vaccines if there is a combined defect). Give prompt antibiotic therapy for breakthrough infection, and consider continuous antibiotic prophylaxis in children with recurrent breakthrough infection despite adequate immunoglobulin troughs. [6]
Step four is lung protection. Establish baseline and repeat annual spirometry in cooperative children, perform low-dose chest computed tomography when bronchiectasis is suspected, and involve physiotherapy and a respiratory team for children with established structural lung disease. The aim is to detect and treat bronchiectasis early, because established bronchiectasis is the single largest determinant of long-term morbidity in antibody deficiency. [7]
Step five is planned transition to adult immunology. A structured handover in adolescence — with documented genotype, vaccination history, lung function trajectory, and an individualised immunoglobulin regimen — preserves continuity and adherence. Genetic counselling for the family, including carrier testing for XLA in maternal relatives, belongs here too. [5] [6]
Specific Subtypes & Scenarios
Each major antibody deficiency carries a distinctive decision point, and a fellowship answer earns depth by handling them individually rather than as a single block. [2]
X-linked agammaglobulinaemia is the archetype. The diagnosis rests on absent B cells, pan-hypogammaglobulinaemia, and a pathogenic BTK variant. Treatment is lifelong immunoglobulin replacement from the time of diagnosis, and carrier testing of maternal female relatives is essential because each carrier son has a one-in-two chance of being affected. The Winkelstein United States registry of 201 patients documented the infection burden and the value of consistent replacement in improving outcomes. [3]
Common variable immunodeficiency demands more than infection management. CVID carries a substantial burden of autoimmune and lymphoproliferative disease and an increased lymphoma risk, so surveillance for immune cytopenias, granulomatous disease, and lymphadenopathy is part of routine care. Two diagnostic criteria frameworks — the Ameratunga criteria and the ESID registry definitions — formalise the diagnosis, and the New Zealand CVID cohort study compared their performance in a real population. Replacement is reserved for those with documented infection susceptibility, not every low IgG. [4] [10] [6]
Specific antibody deficiency is easily missed because total immunoglobulins are normal. The diagnosis depends on a documented failure of pneumococcal polysaccharide vaccine response in a child with recurrent sinopulmonary infection. Most children are managed with antibiotic prophylaxis and prompt treatment of breakthrough infection; immunoglobulin replacement is reserved for those with severe or recurrent infection despite prophylaxis. [2]
Transient hypogammaglobulinaemia of infancy is the diagnosis you make on the way to not treating. The infant has low IgG but thriving growth, preserved vaccine response, and B cells present. Management is expectant: monitor immunoglobulins and clinical status, treat infections as they arise, and reassure the family that the deficit resolves by age three to four years. The harm to avoid is unnecessary, lifelong immunoglobulin therapy in a child who was always going to be normal. [6]
Why over-diagnosis of CVID is harmful
Immunoglobulin replacement is lifelong, costly, and carries infusion-reaction, line, and blood-borne-infection risks. Labelling a child with transient hypogammaglobulinaemia as CVID commits them to decades of unnecessary therapy and to the anxiety of a chronic-immunodeficiency label. The functional vaccine response and the clinical trajectory — not a single low IgG — separate the two. [4] [5]
Complications & Pitfalls
The complications of antibody deficiency divide into the consequences of untreated disease and the consequences of treatment itself, and a fellowship answer handles both. [7] [5]
Bronchiectasis is the dominant long-term complication. Recurrent or undertreated sinopulmonary infection progressively destroys the airway wall, and established bronchiectasis is the strongest predictor of long-term morbidity and mortality. The European Chest CT Group data show that bronchial pathology is common and frequently under-recognised, which is why baseline and surveillance imaging belong in the management plan. [7]
Infection with opportunistic or unusual organisms marks the more severe defects. Chronic enteroviral meningoencephalitis is characteristic of XLA, Pneumocystis pneumonia signals a combined defect or hyper-IgM syndrome, and chronic Giardia or Campylobacter diarrhoea reflects the failure of mucosal antibody. These infections are both a complication and a diagnostic clue. [3] [2]
Autoimmune and lymphoproliferative disease complicate CVID disproportionately. Immune thrombocytopenia, autoimmune haemolytic anaemia, granulomatous disease, and an increased risk of lymphoma mean that surveillance for these is part of routine CVID care, separate from the infection burden. [4]
The pitfalls of treatment are equally important. Infusion reactions to intravenous immunoglobulin, the discomfort and local reactions of subcutaneous immunoglobulin, line-related infection, and the theoretical risk of blood-borne pathogen transmission all weigh against starting therapy without a confirmed indication. The commonest pitfall, however, is over-diagnosis: starting immunoglobulin for a single low IgG in a thriving infant with a self-limiting maturational delay. [5] [6]
Prognosis & Disposition
Prognosis in antibody deficiency is dictated by the underlying entity, the timeliness of diagnosis, and the quality of lung surveillance. With early diagnosis and consistent immunoglobulin replacement, children with XLA survive into adulthood and lead near-normal lives, although they remain dependent on lifelong therapy. [3]
CVID carries a more guarded prognosis because of its autoimmune, granulomatous, and lymphoproliferative complications. Survival is reduced relative to the general population, driven principally by structural lung disease and lymphoma, which is why surveillance and prompt treatment of complications matter as much as the replacement therapy itself. [4] [5]
Transient hypogammaglobulinaemia of infancy has an excellent prognosis: resolution by age three to four years with no long-term sequelae, provided the label is correct and no unnecessary therapy is given. Selective IgA deficiency is similarly benign for most, with only a minority experiencing clinically significant infection or the transfusion-related risk. [6]
Disposition for a general paediatrician is shared care with a clinical immunology service. The immunologist owns the diagnostic characterisation, the immunoglobulin regimen, and the genetic counselling; the general paediatrician owns the acute-infection management, the growth and development surveillance, and the coordination of transition. Early referral to an immunology centre at the point of suspicion — rather than after a confirmed label — is the disposition rule, because the work-up is specialised and the treatment is lifelong. [5] [6]
Special Populations
Antibody deficiency interacts with the child's social and developmental context, and a fellowship answer recognises that the same diagnosis behaves differently across populations. Access, adherence, and late presentation all shape outcome. [6]
Indigenous children, particularly in Australia and New Zealand, face a high background burden of recurrent respiratory infection, crowded housing, and reduced access to specialist services, which both raises the threshold for suspecting a primary defect and lowers the margin for delay when one exists. Structural lung disease is common in these communities, so a confirmed antibody deficiency must be managed intensively to prevent rapid progression to bronchiectasis. [7]
Migrant, refugee, and asylum-seeking families may have incomplete vaccination records, uncertain family history, and barriers to consistent immunoglobulin access. A careful reconstruction of the infection history, confirmation of vaccination status, and a plan that accounts for mobility and language are essential. Interpreter use and trauma-informed communication belong here. [6]
Socioeconomically disadvantaged families carry a disproportionate burden of recurrent infection and late presentation. Subcutaneous immunoglobulin home therapy, where feasible, improves adherence and reduces travel burden, and should be discussed as an option for stable patients. The aim is to fit the therapy to the family's reality rather than the reverse. [5]
Adolescents in transition are a population in their own right. Adherence to immunoglobulin therapy declines in adolescence, the risk-taking that accompanies chronic illness rises, and the handover to adult care is a vulnerable point. A structured, documented transition that preserves continuity of the immunoglobulin regimen and the lung-surveillance trajectory is the safeguard. [5]
Evidence, Guidelines & Regional Differences
The evidence base for antibody deficiency in children rests on three pillars: the IUIS classification that defines the phenotypes, the registry and cohort data that describe outcomes, and the consensus criteria that standardise diagnosis. [2]
The 2022 IUIS update of the phenotypic classification of inborn errors of immunity is the current reference framework for naming and grouping these conditions. It supersedes the 2017 and 2019 versions and reorganises the predominantly antibody deficiencies with updated gene assignments. A fellowship answer should reference the most recent classification rather than an older edition. [2]
Diagnostic criteria for CVID are formalised in two complementary frameworks. The Ameratunga criteria and the European Society for Immunodeficiencies (ESID) registry working definitions specify the immunoglobulin pattern, the functional vaccine response requirement, the age cut-off, and the exclusion of secondary causes. The New Zealand CVID cohort study compared these criteria in a real population and showed that they identify overlapping but not identical groups, which is why both the quantity and the function matter. [4] [10] [6]
Outcome and complication data come from registries and consortia. The Winkelstein XLA registry of 201 patients defined the United States infection burden, and the European Chest CT Group quantified the bronchial pathology that drives long-term morbidity. These data underpin the emphasis on early diagnosis, consistent replacement, and structured lung surveillance. [3] [7]
Regional differences are practical rather than scientific. Australia and New Zealand follow the IUIS and ESID frameworks, with local immunoglobulin product availability and funding arrangements shaping the choice between intravenous and subcutaneous therapy. Indigenous-health and remote-access considerations specific to the region intensify the need for early referral and home-based subcutaneous therapy where appropriate. The warning-sign frameworks (Jeffrey Modell ten signs, modified Düsseldorf criteria) apply universally, but their sensitivity is modest, so clinical judgement remains the final arbiter. [8] [9]
In Australia and New Zealand, suspected primary antibody deficiency is referred to a specialist clinical immunology service for diagnostic characterisation and for the immunoglobulin product, which is funded through national blood arrangements. Subcutaneous home immunoglobulin therapy is increasingly used for stable patients to improve adherence and reduce travel, particularly in remote communities. Always involve the regional immunology centre early — the work-up and lifelong therapy are specialist-led. [5] [6]
Exam Pearls
A fellowship candidate answering on antibody deficiency should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [2] [6]
Anchor one: the functional vaccine response is the decisive test. A low immunoglobulin number alone is not a diagnosis. The pneumococcal polysaccharide response separates a true defect from a number, and it identifies specific antibody deficiency when total IgG is normal. [4]
Anchor two: the B-cell count separates agammaglobulinaemia from hypogammaglobulinaemia. Absent B cells mean XLA or an autosomal recessive mimic and mandate lifelong replacement; B cells present mean CVID, THI, or specific antibody deficiency, each with a different threshold for replacement. [3]
Anchor three: replace immunoglobulin only for confirmed infection susceptibility with a quantified defect. Transient hypogammaglobulinaemia of infancy and many secondary causes do not need replacement, and over-treatment is a recognised harm. [5] [6]
Anchor four: exclude secondary causes before confirming a primary defect. Protein loss, malignancy, immunosuppression, and HIV all produce low immunoglobulin and are often reversible. [6]
Anchor five: protect the lung and plan the transition. Bronchiectasis is the dominant long-term complication, and a structured adolescent handover to adult immunology preserves continuity and adherence. [7] [5]
The three traps to avoid are diagnosing CVID on a single low IgG without functional testing or exclusion of secondary causes, forgetting that immunoglobulin reference ranges are age-adjusted, and giving live vaccines before excluding a combined immunodeficiency. Avoid these and the rest of the answer falls into place. [2] [4]
References
- [1]Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol, 2007.PMID 17577648
- [2]Bousfiha A, Moundir A, Tangye SG, Picard C, Ochs HD, Al-Herz W, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol, 2022.PMID 36198931
- [3]Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore), 2006.PMID 16862044
- [4]Ameratunga R, Woon ST, Brewerton M, Koopmans W, Jordan A, Norton R, et al. Diagnostic criteria for common variable immunodeficiency disorders. J Allergy Clin Immunol Pract, 2016.PMID 27587325
- [5]Bonilla FA. Personalized therapy for common variable immunodeficiency. Allergy Asthma Proc, 2020.PMID 31888779
- [6]Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract, 2019.PMID 30776527
- [7]Schütz K, Fitting C, Hoernes M, Schmidt C, Litzman J, Witte T, et al. Imaging of Bronchial Pathology in Antibody Deficiency: Data from the European Chest CT Group. J Clin Immunol, 2019.PMID 30547383
- [8]Eldeniz FC, Ozdemir E, Kiykim A, Cagdas D, Tezcan I, Ceyhan M, et al. Evaluation of the 10 Warning Signs in Primary and Secondary Immunodeficient Patients. Front Immunol, 2022.PMID 35634313
- [9]Lankisch P, Schiffner J, Backhaus J, Ritzke I, Wirth T, Laws HJ, et al. The Duesseldorf warning signs for primary immunodeficiency: is it time to change the rules? J Clin Immunol, 2015.PMID 25749910
- [10]Ameratunga R, Allan C, Woon ST. Comparison of Diagnostic Criteria for Common Variable Immunodeficiency Disorders (CVID) in the New Zealand CVID Cohort Study. Clin Rev Allergy Immunol, 2021.PMID 34236581