Paeds · allergy-and-immunology
Complement deficiencies
Also known as Complement component deficiency · Classical pathway deficiency · Alternative pathway deficiency · Terminal complement deficiency · Hereditary angioedema due to C1-inhibitor deficiency · Inborn errors of the complement system
A fellowship approach to complement deficiencies in children: recognise the three clinical fingerprints (recurrent invasive Neisseria, unexplained angioedema without urticaria, and childhood-onset lupus-like disease), screen with the total haemolytic complement CH50 and AP50 rather than a lone C3/C4, localise the block to a pathway, and then tailor prevention to the site of the defect — meningococcal vaccination and antibiotic prophylaxis for terminal-pathway loss, C1-inhibitor-directed therapy for hereditary angioedema, and specialist-led surveillance for the immune-complex phenotypes.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A previously well fifteen-year-old presents with his second episode of invasive meningococcal septicaemia in two years. He recovered fully each time and is entirely well between episodes. His C3 and C4 are normal, but his total haemolytic complement (CH50) is undetectable, and the AP50 is normal. Two doors open from that single result. Through one door sits a boy with a terminal complement component deficiency (C5, C6, C7, C8 or C9) who will live a normal life provided he receives meningococcal vaccination, standing antibiotic prophylaxis and a plan for immediate-access emergency antibiotics. Through the other sits a child in whom the low CH50 reflects sample handling or an acquired consumption — and who will be labelled with a chronic immunodeficiency and subjected to years of unnecessary intervention if the result is not repeated and the components quantified. Telling those two children apart — without dismissing the recurrent meningococcaemia as "unlucky" and without over-labelling the well child — is the whole skill of this topic. [9] [10]
C.O.M.P.L.E.M.E.N.T.
Overview & Definition
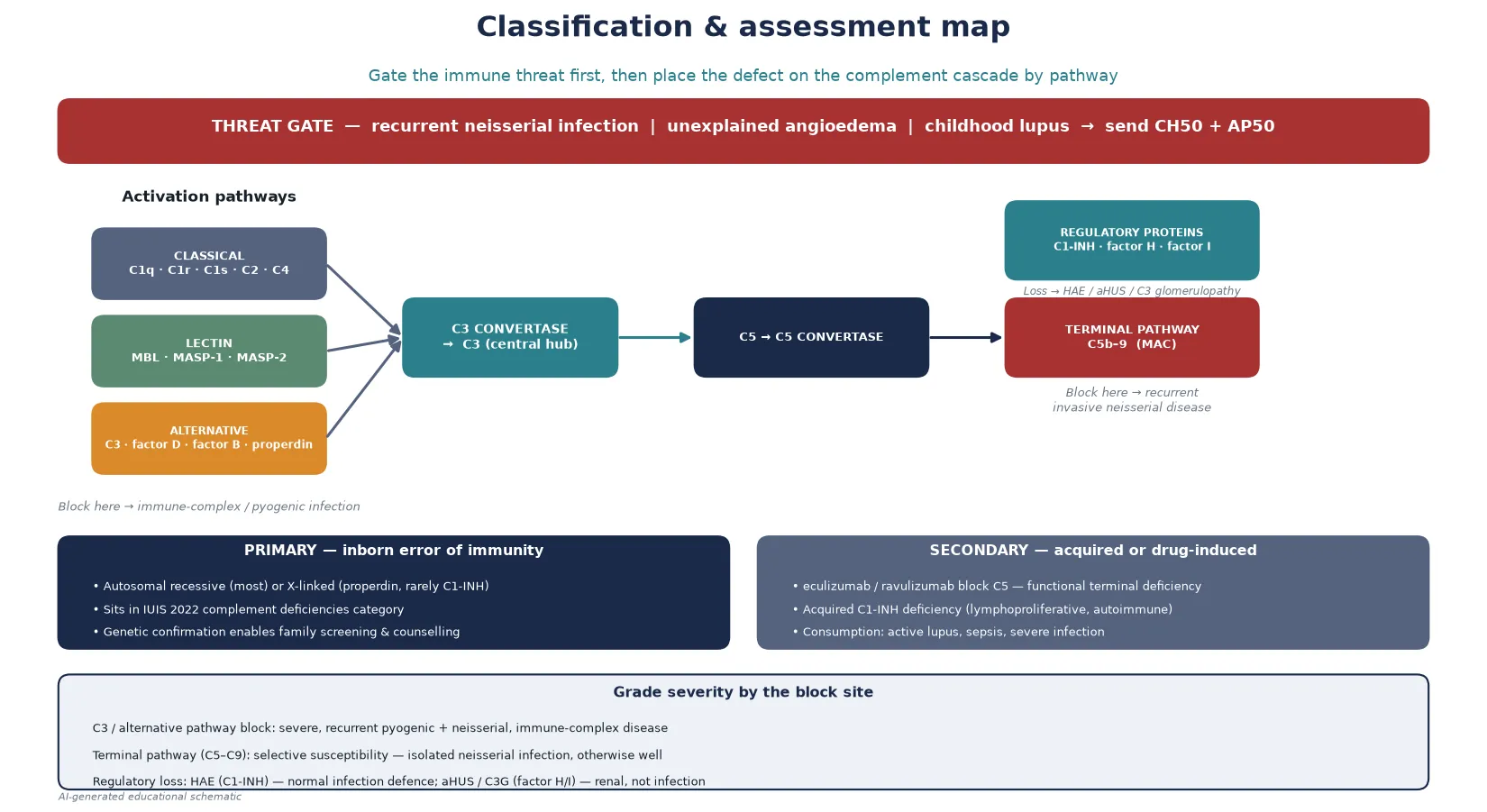
The complement system is a cascade of plasma and cell-surface proteins that, once activated, opsonise pathogens, recruit inflammatory cells, and directly lyse susceptible organisms through the membrane attack complex. Three activation pathways converge on the central component C3: the classical pathway (triggered by antigen–antibody complexes), the lectin pathway (triggered by microbial sugars binding mannose-binding lectin), and the alternative pathway (a continuously ticking surveillance loop). All three generate C3 convertases that cleave C3, and the downstream C5 convertase drives assembly of the terminal membrane attack complex C5b–9. [1] [2]
A complement deficiency is any inborn error or acquired loss that removes a component or a regulator of this cascade. The clinical consequence is not one syndrome but several, and which one a child develops is determined entirely by the site of the block. Loss of an early classical-pathway component impairs clearance of immune complexes and apoptotic debris and predisposes to lupus-like disease. Loss of C3 or an alternative-pathway component compromises opsonisation and produces severe, recurrent pyogenic and neisserial infection. Loss of a terminal component removes the lytic pathway and produces selective, isolated susceptibility to Neisseria. Loss of a regulator produces either uncontrolled bradykinin generation (hereditary angioedema) or uncontrolled C3 activation on the glomerulus (atypical haemolytic uraemic syndrome, C3 glomerulopathy). [3]
The headline distinction for a general paediatrician is that a normal C3 and C4 does not exclude a complement deficiency. The routine immunochemistry assays measure the intact protein concentration, and most component deficiencies leave C3 and C4 in the reference range. The correct screen is the functional haemolytic assay — CH50 for the classical and terminal pathway, AP50 for the alternative pathway — which tests the integrity of the whole cascade by measuring its capacity to lyse sensitised red cells. [1] [6]
Classification
Sort complement deficiency by the site of the block in the cascade, because that single axis predicts the clinical phenotype, the inheritance, and the prevention strategy. The 2022 IUIS classification of inborn errors of immunity groups the complement deficiencies together, and the phenotypes map cleanly onto the cascade anatomy. [5]
Classical pathway deficiencies (C1q, C1r, C1s, C2, C4) impair immune-complex and apoptotic clearance. C1q, C1r, C1s and C4 deficiency carry a very high lifetime risk of lupus-like disease — C1q deficiency in particular is one of the strongest single associations with systemic lupus erythematosus known. C2 deficiency is the commonest classical-pathway defect in European-ancestry populations; it carries a modest lupus risk and some recurrent infection susceptibility, and many affected individuals are entirely asymptomatic. [3] [2]
Lectin pathway deficiencies (mannose-binding lectin, MASPs) are common as polymorphisms but usually produce only a mild increase in recurrent infection in early childhood, and many authorities do not regard isolated low mannose-binding lectin as a disease in its own right. The alternative pathway and C3 sit at the centre: deficiency of C3 itself, factor D, factor B, or properdin compromises opsonisation and produces severe, recurrent pyogenic and neisserial infection together with immune-complex disease. Properdin deficiency is the X-linked exception in a group that is otherwise autosomal recessive, and it classically presents with fulminant meningococcal disease in males. [1] [4]
Terminal pathway deficiencies (C5, C6, C7, C8, C9) remove the lytic membrane attack complex. Their signature is selective, isolated susceptibility to invasive Neisseria — affected children are otherwise entirely well, have no increase in other infections, and may first present after two or more episodes of meningococcal septicaemia. C9 deficiency is particularly common in the Japanese population, where it is often picked up on screening and may be asymptomatic. [9] [10]
Finally, the regulatory deficiencies cut across the anatomy. C1-inhibitor deficiency produces hereditary angioedema — a bradykinin-mediated angioedema with normal infection defence, inherited as an autosomal dominant trait. Loss-of-function in the alternative-pathway regulators factor H, factor I, or membrane cofactor protein produces uncontrolled C3 activation on the glomerular endothelium and presents as atypical haemolytic uraemic syndrome or C3 glomerulopathy. These are renal and thrombotic microangiopathic diseases, not infection susceptibility. [8] [3]
Epidemiology & Risk Factors
Complement deficiencies are individually rare, but the terminal-pathway group is common enough that every general paediatrician will meet recurrent meningococcal disease in a child whose investigation is incomplete. The reported prevalence of terminal complement deficiency varies by population: C5–C9 deficiency is found in roughly one-half to one per cent of patients with a single episode of invasive meningococcal disease, and in a substantially higher fraction of those with recurrent episodes. C9 deficiency is notably common in the Japanese population. [10] [9]
Properdin deficiency is the X-linked member of the group and is important because of its fulminant presentation — affected males carry a high case-fatality from meningococcal disease, and the family history of male deaths from meningitis is the clue that demands investigation of maternal male relatives. C2 deficiency is the commonest classical-pathway defect in European-ancestry populations, linked to the HLA region, but most affected individuals are asymptomatic. C1q, C1r, C1s and C4 deficiencies are rare but carry the strongest lupus associations. [3] [2]
Risk factors are family history and consanguinity. A family history of recurrent meningococcal disease, unexplained male deaths from meningitis, childhood-onset lupus, or known complement deficiency should lower the threshold for sending a CH50 and AP50 in a child who presents with a compatible illness. Population ancestry matters for the specific entities — Japanese ancestry raises the probability of C9 deficiency, and northern European ancestry raises C2 deficiency — but the work-up itself is ancestry-independent. [5]
An increasingly important "risk factor" is iatrogenic: the monoclonal antibodies eculizumab and ravulizumab, used for atypical haemolytic uraemic syndrome, paroxysmal nocturnal haemoglobinuria and some neuromyelitis optica and myasthenia cases, are terminal complement (C5) inhibitors. Any child receiving these agents becomes functionally C5-deficient and carries the same meningococcal risk as an inborn terminal-pathway deficiency — they must be vaccinated and receive antibiotic prophylaxis for the duration of therapy. [10] [8]
Pathophysiology
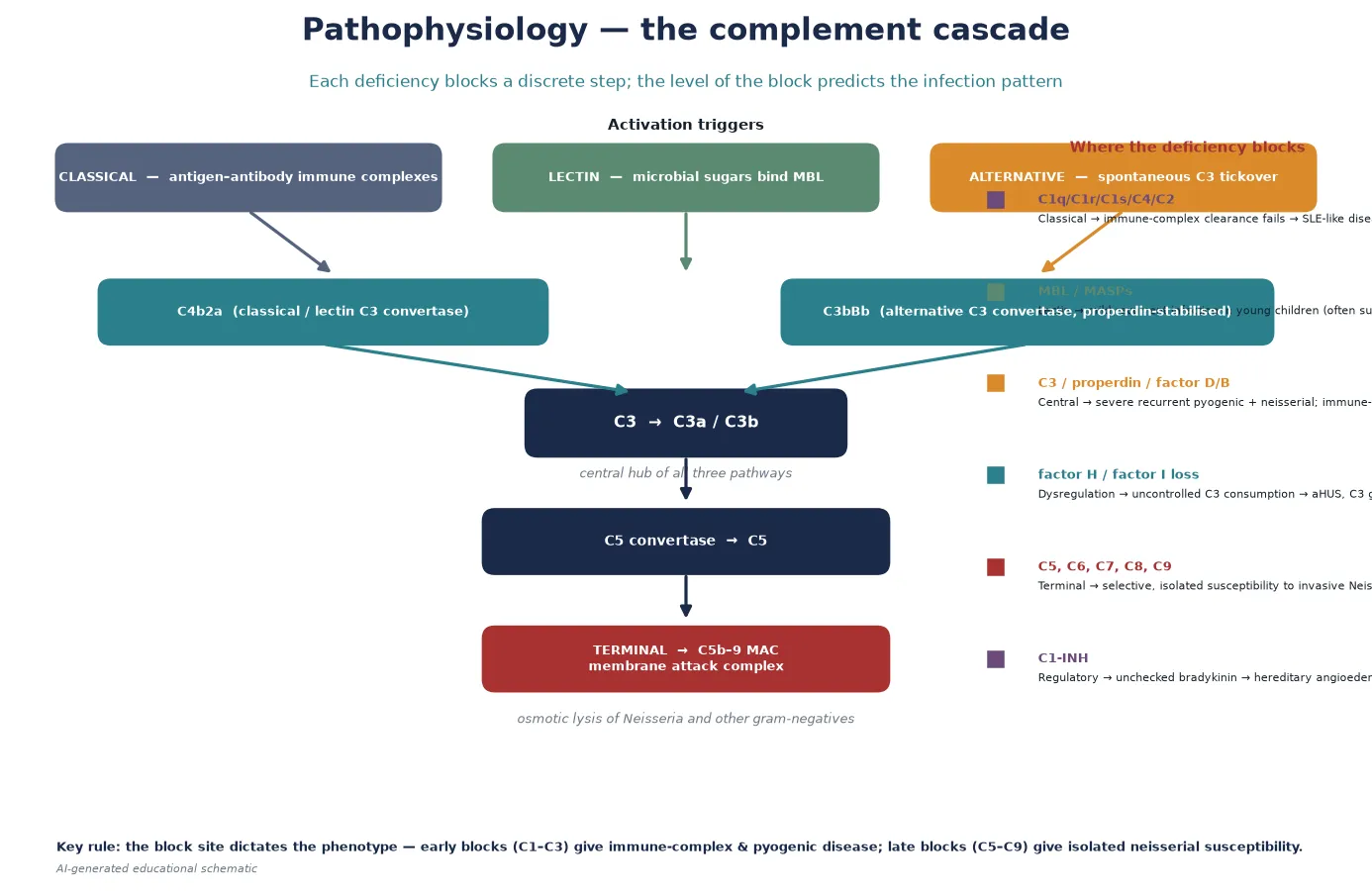
The pathophysiology of complement deficiency is determined by the function of the missing protein, and a fellowship answer earns its depth by linking each block to its downstream consequence. The cascade is best understood as three activation pathways converging on C3, then a single terminal effector arm leading to the membrane attack complex. [1]
Classical pathway blocks remove the system that clears immune complexes and apoptotic cells. C1q binds immune complexes and apoptotic blebs, and C1q-deficient individuals cannot clear this debris; the unprocessed nuclear material drives a type I interferon response and a lupus-like illness. This is why C1q deficiency carries one of the highest disease associations in all of medicine — most affected individuals develop systemic lupus erythematosus, often severe and in childhood. C4 and C2 deficiency share the mechanism but with lower penetrance. [3] [2]
Central C3 and alternative-pathway blocks remove opsonisation. C3b opsonises bacteria for phagocytosis, and properdin stabilises the alternative-pathway C3 convertase that amplifies the response. C3-deficient patients therefore cannot opsonise encapsulated bacteria or clear immune complexes, and they develop severe recurrent pyogenic infection, immune-complex disease, and — as Pekkarinen and colleagues showed — dysregulation of adaptive immune responses, with impaired germinal centre reactions and antibody quality. Properdin and factor D deficiency share the alternative-pathway failure but with a particular susceptibility to meningococcal disease. [4] [1]
Terminal pathway blocks (C5–C9) remove the membrane attack complex. C5b recruits C6, C7, C8 and C9 to assemble a pore-forming complex that inserts into the membrane of susceptible gram-negative organisms — above all Neisseria, which is unusually dependent on complement-mediated lysis because its capsule resists phagocytosis. Without the terminal pathway, Neisseria cannot be killed by serum, and the child develops invasive meningococcal disease. Because opsonisation and immune-complex clearance remain intact, the child has no increase in other infections — the phenotype is selective and isolated. [9] [10]
Regulatory blocks produce dysregulation rather than infection susceptibility. C1-inhibitor restrains the contact (kallikrein–kinin) system as well as the classical and lectin complement pathways; its loss allows uncontrolled bradykinin generation, producing episodic angioedema of the subcutaneous and submucosal tissues. Factor H and factor I restrain the alternative pathway on host cell surfaces; their loss allows uncontrolled C3 activation on the glomerular endothelium, producing atypical haemolytic uraemic syndrome and C3 glomerulopathy. These are thrombotic microangiopathic and renal diseases — the mechanism is over-activity, not deficiency. [8] [3]
Clinical Presentation
Complement deficiency does not have one presentation; it has three clinical fingerprints, and recognising which one a child carries is the first skill. Each fingerprint points to a different part of the cascade. [3]
The first fingerprint is recurrent invasive Neisseria. A child who has had two or more episodes of proven meningococcal meningitis or septicaemia — particularly if they recover fully and are completely well between episodes — has a terminal complement deficiency until shown otherwise. The paradox examiners test is that these children are otherwise well: they do not have recurrent pneumonia, they do not fail to thrive, and their other infection rate is normal. The missing piece is the lytic pathway that Neisseria uniquely depends on. Properdin deficiency shares the neisserial susceptibility but adds fulminance and an X-linked family pattern. [9] [10]
The second fingerprint is unexplained angioedema without urticaria. Recurrent episodes of non-pitting swelling of the skin, gastrointestinal tract (producing colicky abdominal pain and vomiting) or upper airway — without itch, without wheals, and not responding to adrenaline, antihistamines or corticosteroids — is the signature of hereditary angioedema due to C1-inhibitor deficiency. The absence of urticaria is the discriminator from histamine-mediated angioedema. Upper-airway swelling is the life-threatening manifestation, and a family history of similar swelling or unexplained airway events supports the diagnosis. [7] [8]
The third fingerprint is childhood-onset lupus-like disease. A child presenting with systemic lupus erythematosus, or a lupus-like illness with low complement, should be screened for classical pathway deficiency — C1q, C1r, C1s, C2 and C4 deficiency. The lupus is often severe and early-onset, and the family may carry the deficiency. C3 and alternative-pathway deficiency add severe recurrent pyogenic infection — pneumonia, meningitis, septicaemia with encapsulated bacteria — and immune-complex disease such as glomerulonephritis. [3] [2]
The iatrogenic presentation is increasingly common: a child about to commence eculizumab or ravulizumab for atypical haemolytic uraemic syndrome or another indication. They are about to become functionally C5-deficient, and the clinical question is not diagnosis but preparation — meningococcal vaccination and antibiotic prophylaxis before the first dose. [10]
Differential Diagnosis
The differential for each of the three fingerprints is different, and the discipline of this topic is to think structurally about each presentation rather than to chase complement deficiency too early. [6]
For recurrent invasive Neisseria, the alternative explanations are anatomical or functional asplenia (which impairs clearance of encapsulated organisms), antibody deficiency (which raises susceptibility to a broader range of organisms, not just Neisseria), HIV, and simply an epidemiologically heavy exposure in a household or dormitory outbreak. The discriminating feature of terminal complement deficiency is the isolation of the susceptibility — neisserial infection without the broader infection burden that antibody deficiency or asplenia produces. Anatomic asplenia is excluded by examining the spleen and a blood film for Howell-Jolly bodies. [10]
For angioedema without urticaria, the alternatives are histamine-mediated angioedema (which usually has urticaria, itch, and responds to antihistamines), angiotensin-converting enzyme inhibitor–induced angioedema (a drug history is decisive), and acquired C1-inhibitor deficiency (associated with B-cell lymphoproliferative disease or autoimmune disease, typically later-onset and without a family history). The discriminator for hereditary angioedema is the combination of no urticaria, no response to adrenaline, a family history, and onset in childhood or adolescence. [7] [8]
For childhood-onset lupus with low complement, the alternative is classical lupus with immune-complex consumption of C3 and C4 — here the complement is low because of active disease, not because of a deficiency. The discriminator is that consumption normalises when the lupus is treated, whereas a deficiency stays low or absent regardless. Sending a CH50 when C3 and C4 are low from active lupus gives an erroneously low result and can be misleading, so the test is best interpreted away from a flare or repeated once disease is controlled. [3]
For recurrent pyogenic infection broadly, antibody deficiency is the commoner and more important differential, and the two are investigated together — total immunoglobulins and a functional vaccine response sit alongside the CH50 and AP50 in any child with significant infection susceptibility. Secondary complement consumption (active lupus, sepsis, severe infection) lowers C3 and C4 and can lower the CH50, so a single low value in a sick child is not diagnostic and must be repeated when the child is well. [6]
| Fingerprint | Likely alternative | Key discriminator |
|---|---|---|
| Recurrent invasive Neisseria | Anatomic or functional asplenia | Howell-Jolly bodies on film; broader encapsulated-organism susceptibility |
| Angioedema without urticaria | ACE-inhibitor angioedema | Drug history; no family history; resolves on stopping the drug |
| Recurrent pyogenic infection | Antibody deficiency | Broad organism range; low immunoglobulins; poor vaccine response |
Clinical & Bedside Assessment
The bedside task is to decide whether the presentation crosses the threshold for a complement work-up, and to gather the discriminating evidence at the first consultation. Begin with the threat gate: two or more episodes of invasive meningococcal disease, angioedema without urticaria, or childhood-onset lupus with low complement earns a CH50 and AP50. [9] [3]
The history is the discriminating instrument. For suspected terminal-pathway deficiency, establish the number of proven neisserial episodes, the microbiology, the recovery, and crucially whether the child is otherwise well — no recurrent pneumonia, no failure to thrive, no opportunistic infection — because that isolation points to a terminal block. Take a family history for meningitis deaths, particularly in males (properdin), and for consanguinity. For suspected hereditary angioedema, characterise the swelling: site (skin, gut, airway), duration, triggers (trauma, dental work, stress, oestrogen), the absence of urticaria and itch, the lack of response to adrenaline, antihistamines and steroids, and the family history. For suspected classical-pathway deficiency, document the lupus features and the age of onset. [7] [8]
The examination carries fewer specific clues for complement deficiency than for antibody deficiency, but it excludes the mimics. Palpate for splenomegaly or an absent spleen (asplenia mimics terminal deficiency), examine the skin for urticaria (which argues against hereditary angioedema), and look for the stigmata of lupus (malar rash, oral ulcers, arthritis). Plot growth, because failure to thrive with recurrent infection broadens the differential toward antibody or combined deficiency rather than an isolated terminal complement block. Assess the upper airway carefully during any angioedema presentation, because laryngeal involvement is the life-threatening manifestation. [3] [6]
Assess the family's practical context at the same visit. A child in a remote community who develops meningococcal disease faces retrieval delays that make immediate-access emergency antibiotics and a clear action plan more important than for a city-dwelling child. For hereditary angioedema, ensuring the family understands when to use on-demand therapy and when to present to hospital is a bedside task, not a follow-up task. [10]
The decision to investigate rests on the clinical pattern and a correctly chosen screen. A single low C3 and C4 in a sick child reflects consumption and is not diagnostic; a CH50 and AP50 in a well child between episodes is the test that defines a deficiency. Use the threat gate to decide when to send it, and repeat any abnormal result before assigning a label. [1] [6]
Investigations
The investigation ladder is designed to answer a single functional question first: is the whole cascade intact? Quantification with the haemolytic assays comes before identification of the individual component, and genetics confirms the inborn error. [6]
First-line tests are the total haemolytic complement (CH50, also called CH100) and the alternative-pathway haemolytic complement (AP50). The CH50 tests the integrity of the classical and terminal pathways — it is low or absent when any component from C1 through C9 is deficient. The AP50 tests the alternative pathway (C3, factor D, factor B, properdin). A CH50 that is absent or very low with a normal AP50 localises the block to the classical or terminal pathway; an absent AP50 localises it to the alternative pathway. Both assays must be performed on a fresh, properly handled serum sample, because complement is labile and a mishandled sample gives a falsely low result. The routine C3 and C4 are adjunctive — they are usually normal in component deficiencies and low in consumption or classical-pathway deficiency. [1] [2]
Second-line tests identify the missing component. Individual component assays (C1q, C2, C4, C3, C5, C6, C7, C8, C9, properdin, factor D, factor H, factor I) are performed once the haemolytic assay has localised the block. For hereditary angioedema, the decisive tests are C4 (low between and during attacks), C1-inhibitor antigenic level (low in the common type I), and C1-inhibitor functional level (low in both type I and type II). [7] [8]
Genetic testing confirms the inborn error and enables family counselling and cascade screening. Most complement component deficiencies are autosomal recessive; properdin deficiency is X-linked. Next-generation sequencing gene panels are now the standard approach when a primary complement deficiency is suspected. Genetic confirmation is particularly important for properdin deficiency, because it identifies at-risk male relatives who carry a high meningococcal risk, and for the regulatory deficiencies, where the result guides prognosis and family screening. [5]
Autoantibody and functional studies apply to specific scenarios. Anti-C1-inhibitor antibodies and low C1q distinguish acquired from hereditary C1-inhibitor deficiency. Anti-factor H autoantibodies are sought in atypical haemolytic uraemic syndrome. In a child on eculizumab or ravulizumab, no special test is needed to "diagnose" the functional deficiency — it is iatrogenic and known — but meningococcal vaccination status and antibiotic prophylaxis must be confirmed before each cycle. [8] [10]
Management — Resuscitation
Resuscitation in complement deficiency means treating the acute presentation safely while avoiding the two harms specific to this group: giving adrenaline, antihistamines and corticosteroids for hereditary angioedema (they do not work), and dismissing recurrent meningococcal disease as a chance event without investigation. [8] [10]
A child presenting with acute meningococcal sepsis receives the standard paediatric sepsis pathway: prompt cultures, empirical intravenous cefotaxime or ceftriaxone, and fluid, oxygen and inotropic support as required. There is no complement-specific acute therapy for the infection itself — the deficiency is in host defence, and the treatment is standard antimicrobial and supportive care. The complement-specific task is to ensure that, after recovery, the child is investigated with a CH50 and AP50 rather than discharged as a single unlucky event. [9] [10]
The complement-specific resuscitation hazard is treating hereditary angioedema with the wrong drugs. Acute angioedema in C1-inhibitor deficiency is bradykinin-mediated, and adrenaline, antihistamines and corticosteroids — the standard anaphylaxis regimen — are ineffective. The correct acute therapies are intravenous or subcutaneous C1-inhibitor concentrate, the bradykinin B2-receptor antagonist icatibant, or ecallantide (a kallikrein inhibitor), depending on availability and local protocol. If upper-airway obstruction is present or impending, secure the airway early and give the specific therapy. A child with known hereditary angioedema should carry on-demand therapy and an action plan. [7] [8]
The second resuscitation task is preparing a child for a complement inhibitor. Before the first dose of eculizumab or ravulizumab, the child must receive meningococcal ACWY and B vaccination and start antibiotic prophylaxis, because the drug renders them functionally C5-deficient for the duration of therapy. Missing this preparation has caused fatal meningococcal disease. [10]
Resuscitation of the family's understanding matters equally. A child newly diagnosed with a complement deficiency will need a structured plan — vaccines, prophylaxis, an emergency antibiotic supply, and family screening — and parents benefit from an early, honest explanation of what the diagnosis means, what the treatment involves, and what the prognosis is, before the detail of long-term management is introduced. [8] [6]
Management — Definitive & Stepwise
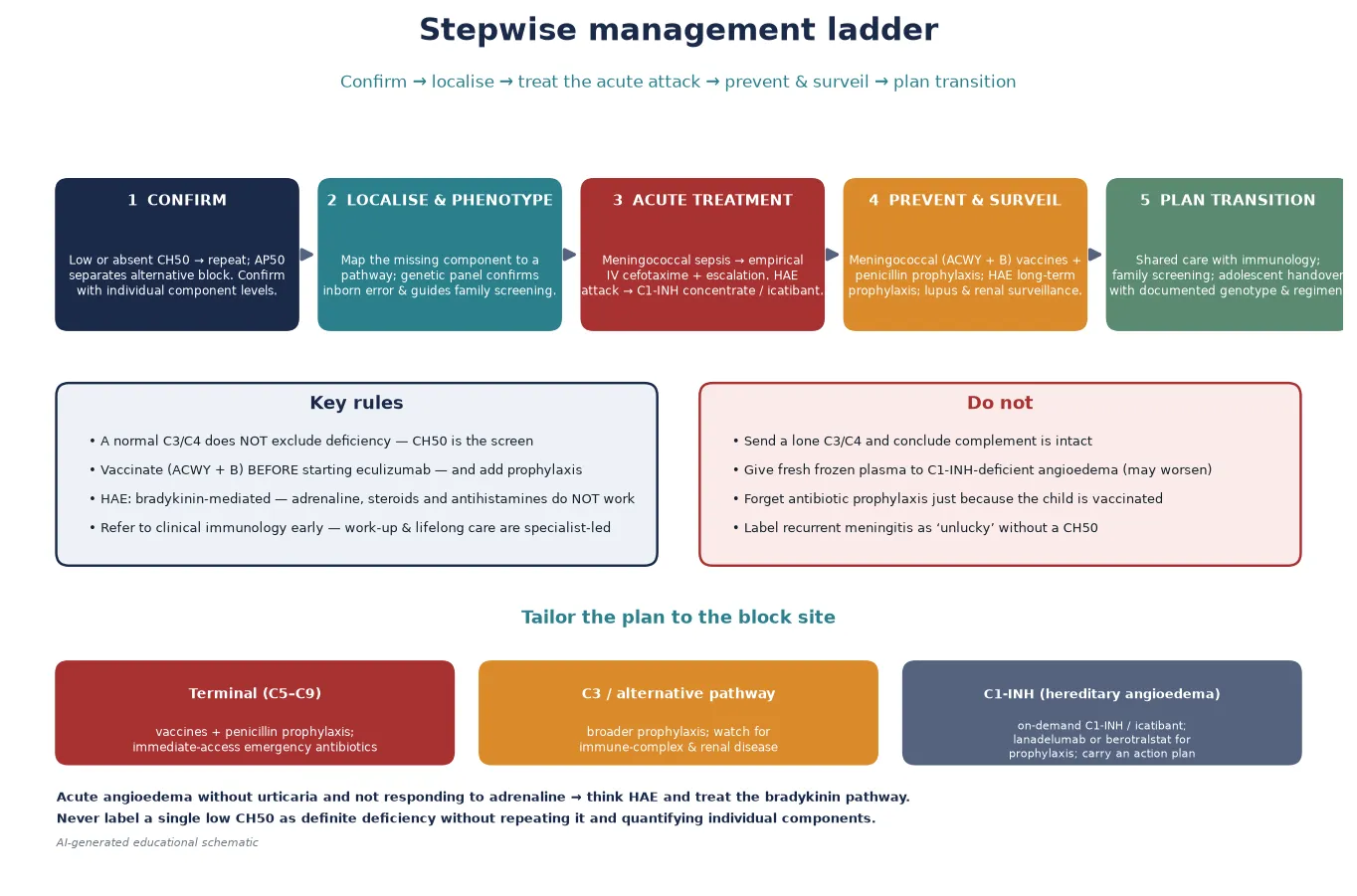
Definitive management follows a five-step ladder that a fellowship candidate can recite: confirm the defect, localise and phenotype, treat the acute attack, prevent and surveil, and plan the transition. The discipline is to tailor prevention to the site of the block. [8] [6]
Step one is to confirm the defect before treating it. A single low CH50 in a sick child often reflects consumption or sample handling. Confirm the defect by repeating the haemolytic assay on a fresh sample when the child is well, then quantify the individual components and confirm genetically. Only a child with a confirmed, quantified deficiency moves to the long-term prevention plan. [1] [6]
Step two is meningococcal vaccination and antibiotic prophylaxis for the infection-defence blocks. Children with terminal-pathway (C5–C9), properdin, factor D, factor B, or C3 deficiency, and any child on eculizumab or ravulizumab, should receive meningococcal ACWY conjugate vaccination, meningococcal B vaccination, and continuous antibiotic prophylaxis (typically penicillin, with consideration of an alternative if colonised). They should also carry a supply of emergency antibiotics for immediate self-administration at the onset of a febrile illness, with clear instructions to present to hospital. Ladhani and colleagues' case series documented the substantial meningococcal burden in complement-deficient patients and underpin this emphasis on prevention. [10] [9]
Step three is pathway-specific therapy for hereditary angioedema. The 2021 WAO/EAACI guideline defines the modern approach: on-demand therapy for acute attacks (C1-inhibitor concentrate or icatibant, carried by the patient), short-term prophylaxis before procedures (C1-inhibitor concentrate), and long-term prophylaxis for patients with frequent or severe attacks (lanadelumab, a kallikrein inhibitor, or berotralstat, an oral plasma kallikrein inhibitor, or C1-inhibitor concentrate). Attenuated androgens are now used less often in children because of their adverse-effect profile. Every patient needs a written action plan and on-demand therapy available at all times. [8] [7]
Step four is surveillance for the immune-complex and renal phenotypes. Children with classical-pathway deficiency need monitoring for lupus-like disease, with urinalysis, blood pressure, and clinical review. Children with factor H or factor I deficiency, or C3 deficiency, need renal surveillance for glomerulonephritis and atypical haemolytic uraemic syndrome — urinalysis, blood pressure, renal function, and full blood count for haemolysis and thrombocytopenia. C3-deficient patients, as Pekkarinen showed, also have dysregulated adaptive immunity and may need antibody-response monitoring. [3] [4]
Step five is planned transition and family screening. Genetic confirmation enables cascade screening of at-risk relatives — particularly important for properdin deficiency (maternal male relatives) and for the autosomal dominant hereditary angioedema. A structured adolescent handover to adult immunology, with documented genotype, vaccination and prophylaxis status, and an action plan, preserves continuity. [5] [8]
Specific Subtypes & Scenarios
Each major complement deficiency carries a distinctive decision point, and a fellowship answer earns depth by handling them individually rather than as a single block. [3]
Terminal pathway deficiencies (C5–C9) are the archetype for the general paediatrician. The child presents with recurrent invasive meningococcal disease and is otherwise completely well. The CH50 is absent or very low; the AP50 is normal unless C3 is involved. Rauscher and colleagues' case discussion of C6 deficiency illustrates the diagnostic journey and the need to avoid dismissing recurrent meningococcaemia as chance. Management is meningococcal ACWY and B vaccination, continuous antibiotic prophylaxis, and an emergency antibiotic supply. The prognosis with consistent prevention is good, and most children live normal lives. [9] [10]
Properdin deficiency is the X-linked exception. It classically presents with fulminant meningococcal disease in males, often with a family history of male deaths from meningitis. The CH50 may be normal or low, and the AP50 is low; the diagnosis requires properdin assay and genetic confirmation. The fulminance and the X-linked inheritance make family screening of maternal male relatives essential — each at-risk male carries a high meningococcal risk. [1] [3]
C3 deficiency is the most severe of the infection-defence blocks. Without C3, opsonisation fails, and the child develops severe recurrent pyogenic infection, immune-complex disease, and — as Pekkarinen and colleagues demonstrated — dysregulated adaptive immunity with impaired antibody responses. Management is broader than the terminal pathway: it combines the meningococcal and antibiotic prophylaxis with surveillance for immune-complex disease and attention to vaccine responses. [4]
Hereditary angioedema (C1-inhibitor deficiency) is the bradykinin phenotype. Type I (85 per cent) has low antigenic and functional C1-inhibitor; type II has normal or high antigenic level but low function. The C4 is characteristically low between and during attacks. The 2021 WAO/EAACI guideline defines on-demand therapy (C1-inhibitor concentrate or icatibant), short-term prophylaxis before procedures, and long-term prophylaxis (lanadelumab, berotralstat, or C1-inhibitor). The life-saving message is that acute attacks do not respond to adrenaline, antihistamines or corticosteroids — the wrong drugs are still the commonest error. [7] [8]
Classical pathway deficiency and childhood lupus. C1q deficiency carries the strongest lupus association; C4 and C2 carry lower-penetrance risk. A child with lupus and persistently low complement despite disease control should be screened for a classical-pathway deficiency. C2 deficiency, the commonest, is often asymptomatic and may be found incidentally. [3] [2]
Why treating hereditary angioedema with anaphylaxis drugs is harmful
Hereditary angioedema is bradykinin-mediated, not histamine-mediated. Adrenaline, antihistamines and corticosteroids do not abort the attack, and relying on them delays the only effective therapy (C1-inhibitor concentrate or icatibant) — a dangerous delay when the upper airway is swelling. The absence of urticaria and itch, and the failure to respond to anaphylaxis therapy, are the clues that redirect you to the bradykinin pathway. [7] [8]
Complications & Pitfalls
The complications of complement deficiency divide into the consequences of untreated disease and the consequences of mistreating it, and a fellowship answer handles both. [10] [8]
Invasive meningococcal disease is the dominant complication of the terminal-pathway and alternative-pathway deficiencies. Even with vaccination and prophylaxis, the risk is higher than in the general population, and breakthrough infection may be fulminant. The case-fatality in properdin deficiency is particularly high. The protection against meningococcal disease is imperfect because vaccination does not cover all serogroups and antibody titres wane — which is why antibiotic prophylaxis and an emergency antibiotic supply are added, not substituted, for vaccination. [9] [10]
Lupus and immune-complex disease complicate the classical-pathway and C3 deficiencies. The lupus is often severe and early-onset, and glomerulonephritis may accompany it. For the regulatory deficiencies (factor H, factor I), the complication is atypical haemolytic uraemic syndrome or C3 glomerulopathy — thrombotic microangiopathy and progressive renal disease that require pathway-specific therapy. [3]
Airway compromise is the life-threatening complication of hereditary angioedema. Laryngeal oedema can asphyxiate, and historically a substantial fraction of untreated affected individuals died from airway obstruction. Modern on-demand therapy and action plans have transformed this prognosis, but only if the patient carries and uses the therapy. [7] [8]
The pitfalls of management are equally important. Treating hereditary angioedema with anaphylaxis drugs is the commonest and most dangerous error. Sending a lone C3 and C4 and concluding that complement is intact is the commonest diagnostic error — the CH50 is the screen. Starting eculizumab without prior vaccination and prophylaxis is an avoidable cause of fatal meningococcal disease. Dismissing recurrent meningococcal disease as "unlucky" without a CH50 is the error that delays diagnosis of a treatable deficiency. And a single low CH50 in a sick child is often consumption, not deficiency — repeat it before labelling. [1] [6]
Prognosis & Disposition
Prognosis in complement deficiency is dictated by the entity, the timeliness of diagnosis, and the consistency of prevention. Children with terminal-pathway deficiency who receive vaccination, prophylaxis and an emergency antibiotic plan have a good prognosis — the meningococcal risk is reduced but not eliminated, and most live normal lives. Properdin deficiency carries a higher fulminance and case-fatality, demanding earlier family screening. [9] [10]
C3 and alternative-pathway deficiency carry a more guarded prognosis because of the combination of severe infection susceptibility, immune-complex disease, and — for the regulatory defects — renal disease. C1q deficiency has a poor prognosis driven by severe childhood lupus. Hereditary angioedema has an excellent prognosis with modern on-demand and prophylactic therapy, but a dangerous prognosis without it, because of airway compromise. [3] [7]
Disposition for a general paediatrician is shared care with a clinical immunology service. The immunologist owns the diagnostic characterisation, the genetic counselling, and the pathway-specific therapy (particularly for hereditary angioedema and the glomerulopathic deficiencies); the general paediatrician owns the acute-infection management, the vaccination and prophylaxis logistics, and the coordination of transition. Early referral to an immunology centre at the point of suspicion — rather than after a confirmed label — is the disposition rule, because the work-up is specialised and the prevention is lifelong. [6] [8]
Special Populations
Complement deficiency interacts with the child's social and developmental context, and a fellowship answer recognises that the same diagnosis behaves differently across populations. Access, adherence, and late presentation all shape outcome. [10]
Indigenous children, particularly in Australia and New Zealand, face a high background burden of bacterial respiratory infection and reduced access to specialist services, which both raises the threshold for suspecting a primary defect and lowers the margin for delay when one exists. A confirmed terminal complement deficiency in a remote community demands an even stronger emphasis on immediate-access emergency antibiotics and a clear retrieval plan, because distance converts a manageable infection into a life-threatening one. [9]
Migrant, refugee, and asylum-seeking families may have incomplete vaccination records, uncertain family history (particularly for the X-linked and autosomal dominant conditions), and barriers to consistent specialist access. A careful reconstruction of the infection and family history, confirmation of meningococcal vaccination status, and a plan that accounts for mobility and language are essential. Interpreter use and trauma-informed communication belong here. [6]
Socioeconomically disadvantaged families carry a disproportionate burden of late presentation and may face cost barriers to the self-carried on-demand therapies for hereditary angioedema. Ensuring access to subsidised therapy, written action plans in the family's language, and a clear line to specialist advice is the safeguard. [8]
Adolescents in transition are a population in their own right. Adherence to antibiotic prophylaxis and to on-demand angioedema therapy declines in adolescence, the risk-taking that accompanies chronic illness rises, and the handover to adult care is a vulnerable point. A structured, documented transition that preserves continuity of the prevention regimen and the action plan is the protection. [8] [5]
Evidence, Guidelines & Regional Differences
The evidence base for complement deficiency in children rests on four pillars: the cascade biology defined by the Walport reviews, the IUIS classification that groups the inborn errors, the cohort and case series data that describe the meningococcal burden, and the consensus guidelines that standardise hereditary angioedema management. [1] [5]
The Walport reviews in the New England Journal of Medicine remain the canonical description of the complement cascade — the first part covers activation and the classical and lectin pathways, the second the alternative pathway, regulation, and the biological consequences of deficiency. They are the reference a fellowship answer is built on for the cascade anatomy. [1] [2]
The 2022 IUIS update of the phenotypic classification of inborn errors of immunity groups the complement deficiencies and is the current reference framework for naming and grouping them. The ESID registry working definitions standardise the clinical diagnosis of inborn errors of immunity, including the complement group. A fellowship answer should reference the most recent classification. [5] [6]
The meningococcal burden data come from case series and surveillance. Ladhani and colleagues' case series of invasive meningococcal disease in patients with complement deficiencies documented the substantial burden and the variety of deficiencies represented, and it underpins the emphasis on vaccination and prophylaxis. Rauscher and colleagues' discussion of C6 deficiency illustrates the diagnostic and management approach to a terminal-pathway defect. [10] [9]
The hereditary angioedema guideline is the 2021 WAO/EAACI revision, which defines on-demand therapy, short-term prophylaxis, and long-term prophylaxis with the modern agents (lanadelumab, berotralstat). Zuraw's NEJM clinical practice review remains the accessible reference for the condition. [8] [7]
Regional differences are practical rather than scientific. Australia and New Zealand follow the IUIS, ESID, and WAO/EAACI frameworks, with local immunoglobulin and C1-inhibitor product availability shaping therapy choices. Indigenous-health and remote-access considerations intensify the need for early referral, immediate-access emergency antibiotics, and clear retrieval plans. The Japanese-population predominance of C9 deficiency is a recognised regional pattern. Eculizumab use in atypical haemolytic uraemic syndrome is universal across regions and carries the same vaccination and prophylaxis obligation everywhere. [10] [9]
In Australia and New Zealand, suspected complement deficiency is referred to a specialist clinical immunology service for diagnostic characterisation and for the pathway-specific therapy. Meningococcal ACWY and B vaccination, continuous antibiotic prophylaxis, and a self-carried emergency antibiotic supply are standard for terminal and alternative-pathway deficiencies. Hereditary angioedema therapy (C1-inhibitor concentrate, icatibant, lanadelumab, berotralstat) is available through specialist services with funding arrangements. Indigenous-health and remote-access considerations drive a lower threshold for early referral and stronger immediate-access planning. [8] [10]
Exam Pearls
A fellowship candidate answering on complement deficiency should land five anchor points and avoid four classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [1] [8]
Anchor one: the three clinical fingerprints. Recurrent invasive Neisseria (terminal block), angioedema without urticaria (C1-inhibitor), and childhood-onset lupus with low complement (classical pathway) are the presentations that earn a CH50. Recite them and the work-up follows. [9] [3]
Anchor two: the CH50 is the screen, not the C3/C4. A normal C3 and C4 does not exclude a complement deficiency. The functional haemolytic assay tests the integrity of the whole cascade, and the AP50 separates the alternative pathway. [1] [6]
Anchor three: the block site dictates the phenotype and the prevention. Terminal = isolated neisseria; C3/alternative = severe pyogenic and neisserial; classical = lupus; regulatory = angioedema or glomerular disease. Prevention is meningococcal vaccination and prophylaxis for the infection blocks, pathway-specific therapy for the rest. [9] [8]
Anchor four: hereditary angioedema is bradykinin-mediated. Adrenaline, antihistamines and corticosteroids do not work; C1-inhibitor concentrate or icatibant does. The absence of urticaria and the lack of response to anaphylaxis drugs redirect you. [7] [8]
Anchor five: eculizumab makes a patient functionally C5-deficient. Vaccinate and give prophylaxis before the first dose. This iatrogenic deficiency is increasingly common and examinable. [10]
The four traps to avoid are sending a lone C3/C4 and concluding complement is intact, treating hereditary angioedema with anaphylaxis drugs, starting eculizumab without vaccination and prophylaxis, and dismissing "otherwise well" recurrent meningococcal disease as unlucky. Avoid these and the rest of the answer falls into place. [9] [1]
References
- [1]Walport MJ. Complement. First of two parts. N Engl J Med, 2001.PMID 11287977
- [2]Walport MJ. Complement. Second of two parts. N Engl J Med, 2001.PMID 11297706
- [3]Botto M, Kirschfink M, Macor P, Ciciri F, Debreczeni IF, Fischer E, et al. Complement in human diseases: Lessons from complement deficiencies. Mol Immunol, 2009.PMID 19481265
- [4]Pekkarinen PT, Heikkilä N, Kisand K, Tubrik AL, Lumi T, Rannikmäe K, et al. Dysregulation of adaptive immune responses in complement C3-deficient patients. Eur J Immunol, 2015.PMID 25446578
- [5]Bousfiha A, Moundir A, Tangye SG, Picard C, Ochs HD, Al-Herz W, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol, 2022.PMID 36198931
- [6]Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract, 2019.PMID 30776527
- [7]Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med, 2008.PMID 18768946
- [8]Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema - The 2021 revision and update. World Allergy Organ J, 2022.PMID 35497649
- [9]Rauscher CK, Fajt ML, Bryk JA, Banerji AS, Schneider LC, Hornik CP, et al. Clinical implications of C6 complement component deficiency. Allergy Asthma Proc, 2020.PMID 32867893
- [10]Ladhani SN, Campbell H, Lucidarme J, Ramsay ME, Borrow R, Parikh SR, et al. Invasive meningococcal disease in patients with complement deficiencies: a case series (2008-2017). BMC Infect Dis, 2019.PMID 31200658