Paeds · allergy-and-immunology
Immune dysregulation, lymphoproliferation and autoinflammatory disease
Also known as Inborn errors of immunity · Immune dysregulation disorders · Autoinflammatory syndromes · Periodic fever syndromes · Immune regulatory disorders · ALPS
A fellowship approach to the child with monogenic immune dysregulation, lymphoproliferation or autoinflammatory disease: sort the presentation by its dominant mechanism (failed tolerance, failed apoptosis, or innate cytokine over-activation), clear the haemophagocytic lymphohistiocytosis and macrophage activation syndrome threat gate first, name the canonical diseases and genes, and match therapy to the pathway (IL-1 blockade, colchicine, sirolimus, CTLA4-Ig, JAK inhibition) with haematopoietic stem cell transplant reserved for severe disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Picture three children in a single clinic. An infant with bloody diarrhoea, severe eczema and new type-1 diabetes. A four-year-old with big, painless glands and a low platelet count that keeps dropping. A two-year-old with clockwork fevers every five weeks, a red throat and mouth ulcers, perfectly well between attacks. Each looks like a different problem — a gut disease, a blood disease, a fever mystery. They are not. Each is a single control-point of the immune system failing, and the framework on this page is built so a registrar can sort all three into the right lane and know what to do first. [1] [2]
Three control-points, three failures
Overview & Definition
Immune dysregulation, lymphoproliferation and autoinflammatory disease are grouped in the International Union of Immunological Societies classification as inborn errors of immunity in which the problem is not susceptibility to infection (the classical immunodeficiency) but a loss of the controls that keep the immune response aimed, sized and switched off. Picard and the IUIS committee placed them in the immune-dysregulation and autoinflammatory groups of the 2015 classification, and Tangye and colleagues' 2022 update expanded the catalogue substantially as new genes were identified. [1] [2]
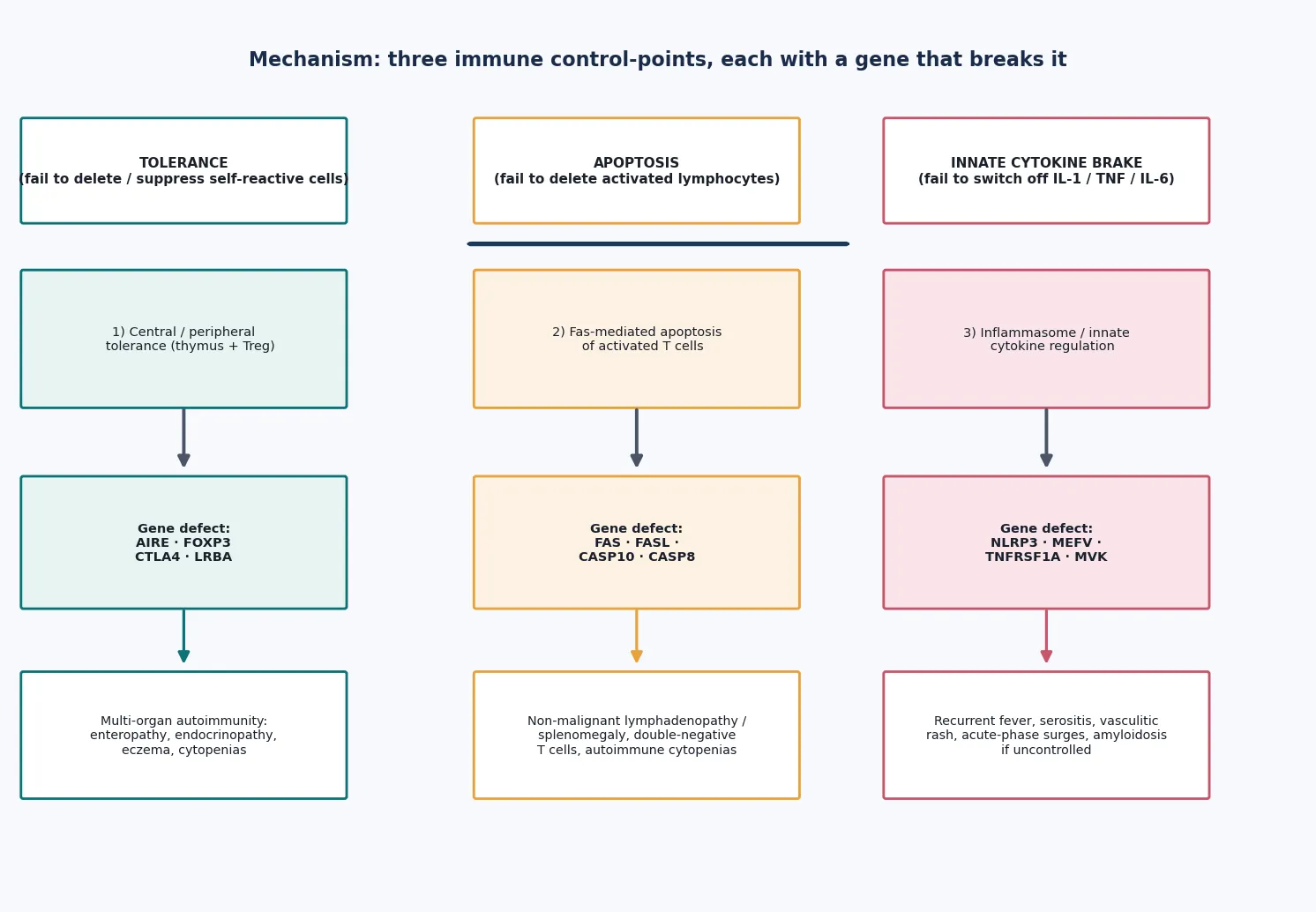
The unifying idea is mechanistic. A normal immune response has to (1) avoid attacking self, (2) contract back down once a threat is cleared, and (3) wind up and down the innate inflammatory engine in proportion to danger. Each of those steps is held in place by specific genes. When a gene in step one breaks, self-tolerance fails and the child develops organ-specific autoimmunity — that is immune dysregulation. When a gene in step two breaks, activated lymphocytes fail to die and accumulate — that is lymphoproliferation. When a gene in step three breaks, innate cytokines such as interleukin-1 surge without provocation and recur — that is autoinflammation. [2]
The distinction between autoimmunity and autoinflammation is worth holding onto even when it blurs. Autoimmunity is adaptive and self-antigen-driven — T cells and antibodies attack a defined target, which is why you find autoantibodies and organ-specific damage. Autoinflammation is innate and largely antigen-independent — it is the inflammasome and cytokine machinery misfiring, which is why attacks recur without infection and why acute-phase markers swing. CTLA4 and LRBA deficiency show why the boundary is porous: a single gene can produce both lymphoproliferation and organ-specific autoimmunity at once. [3]
Classification
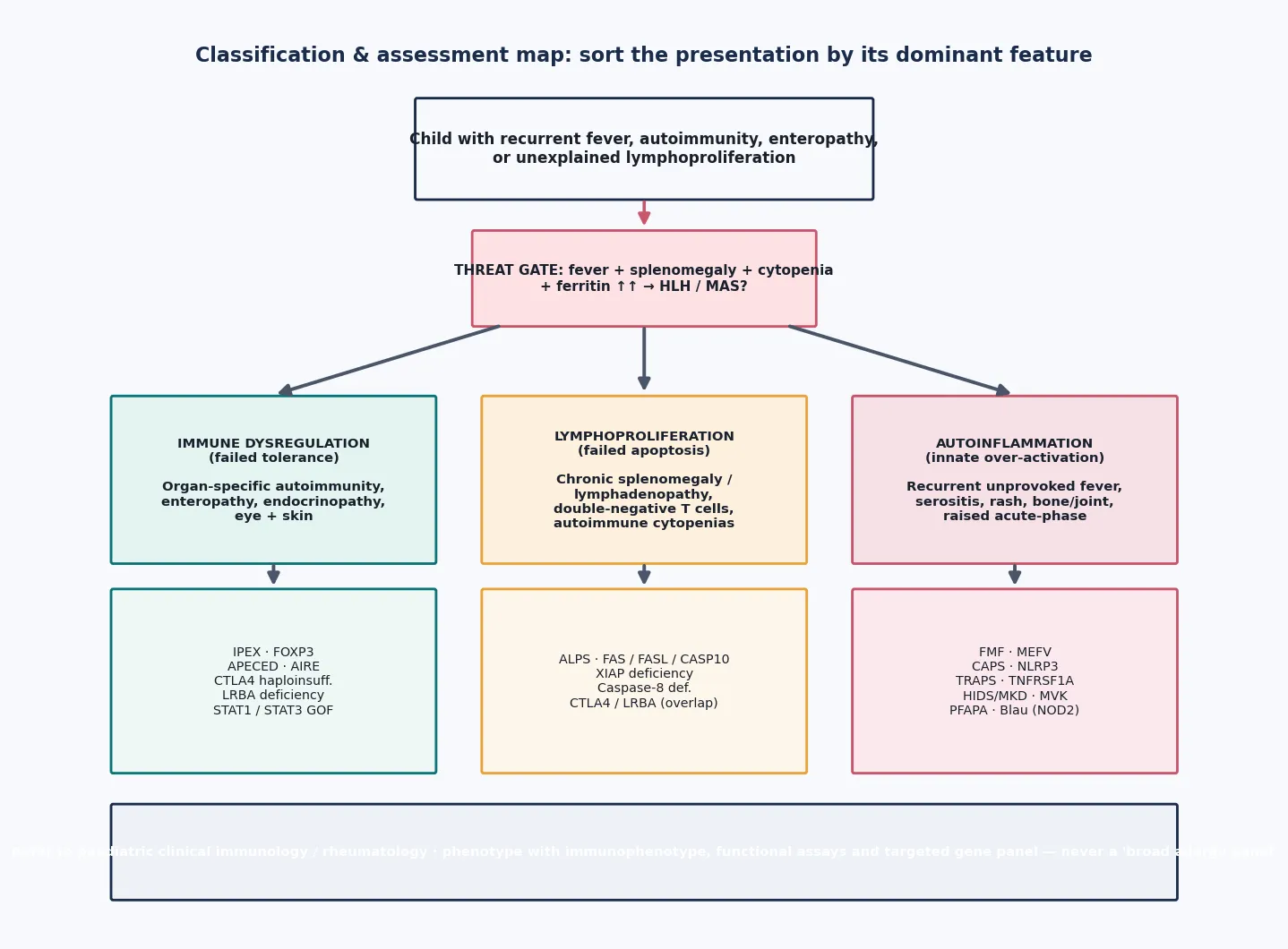
Sort these disorders by the dominant control-point that fails, because that single split predicts the presentation, the test that helps, the drug that works, and whether a transplant is on the table. [1]
Immune dysregulation is tolerance failure. The canonical diseases are IPEX (FOXP3), the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome known as APECED or APS-1 (AIRE), and the regulatory brake-loss disorders CTLA4 haploinsufficiency and LRBA deficiency. The phenotype is early-onset, organ-specific autoimmunity: enteropathy, type-1 diabetes, thyroiditis, eczema, cytopenias and chronic mucocutaneous candidiasis. [3] [4]
Lymphoproliferation is apoptosis failure. The prototype is autoimmune lymphoproliferative syndrome (ALPS), caused by defects in the Fas (FAS), Fas-ligand (FASL) or caspase-10 (CASP10) pathway that prevent activated lymphocytes from dying. The child accumulates chronic, non-malignant lymphadenopathy and splenomegaly, carries a population of double-negative T cells, and develops autoimmune cytopenias. [6] [7]
Autoinflammation is innate cytokine over-activation. The hereditary recurrent fevers dominate: familial Mediterranean fever (MEFV), the cryopyrin-associated periodic syndromes (NLRP3), tumour necrosis factor receptor-associated periodic syndrome (TRAPS, TNFRSF1A), hyper-IgD with periodic fever syndrome also called mevalonate kinase deficiency (MVK), and PFAPA. Blau syndrome (NOD2) adds a granulomatous phenotype. [11] [12]
Epidemiology & Risk Factors
Each of these diseases is rare on its own, but together they form a meaningful diagnostic category that every general paediatrician meets — a child with early-onset, severe or therapy-resistant autoimmunity, unexplained lymphadenopathy, or recurrent unprovoked fevers. The IUIS classification has grown rapidly because next-generation sequencing keeps unmasking new monogenic causes, so the practical message is that the list is always expanding and a phenotype-guided gene panel outperforms memory of a fixed list. [1] [2]
The strongest risk markers are a family history of early-onset autoimmunity, an unexplained childhood death, or parental consanguinity. Many of these disorders are autosomal recessive or X-linked, so a careful pedigree — drawn, not described — changes the prior probability. A founder mutation in a particular community can make a disease common locally that is vanishingly rare elsewhere. [4]
The single most important personal risk factor is severity and timing that exceed the common explanation. Eczema in an infant is common; eczema with intractable bloody diarrhoea and new type-1 diabetes is not. Recurrent tonsillitis is common; clockwork fevers every five weeks with complete wellbeing between attacks is not. Benign cervical lymphadenopathy is common; chronic painless generalised lymphadenopathy with a falling platelet count is not. Severity, multiplicity and therapy-resistance are the flags that should pull a monogenic cause onto the differential. [6] [13]
Pathophysiology
Each of the three mechanisms maps to a single immune control-point with a predictable clinical signature, and that mapping is the reason a targeted therapy works. [2]
In immune dysregulation, tolerance fails. In the thymus, the AIRE protein displays self-proteins to developing T cells so that self-reactive clones are deleted; loss of AIRE (APECED) lets self-reactive cells escape into the periphery. Out in the tissues, FOXP3-positive regulatory T cells suppress autoreactive effector cells; loss of FOXP3 (IPEX) removes that brake. The CTLA4 molecule strips co-stimulatory signals off antigen-presenting cells, so CTLA4 haploinsufficiency or LRBA deficiency releases T cells from regulation. The shared downstream result is organ-specific autoimmunity driven by self-reactive lymphocytes. [3] [4] [5]
In lymphoproliferation, apoptosis fails. After an immune response, activated T cells are meant to die back through the Fas (CD95) receptor and its downstream caspase cascade. When FAS, FASL or CASP10 is defective, those activated cells survive and accumulate — including a distinctive population of mature T cells that carry the alpha-beta T-cell receptor but lack both CD4 and CD8, the double-negative T cells that are the laboratory hallmark of ALPS. These cells are otherwise normal, so the proliferation is non-malignant, but it drives chronic splenomegaly, lymphadenopathy and autoimmune destruction of blood cells. [6] [7]
In autoinflammation, the innate cytokine brake fails. The NLRP3 inflammasome is a molecular platform that activates interleukin-1; gain-of-function variants (the cryopyrin-associated periodic syndromes) leave it switched on. The mevalonate pathway defect in MKD and the aberrant TNF signalling in TRAPS converge on the same endpoint — recurrent, unprovoked surges of innate cytokines, principally interleukin-1, that produce fever, serositis and rash without any infection. This is why blockade of interleukin-1 produces such dramatic responses across the hereditary recurrent fevers. [11] [12]
Clinical Presentation
The three mechanisms produce three recognisable clinical signatures, and recognising the signature is faster than working through a gene list. [2]
Immune dysregulation presents as early-onset, organ-specific autoimmunity that is unusually severe or multi-organ. IPEX classically brings infantile enteropathy with bloody diarrhoea, severe eczema, and early type-1 diabetes or thyroiditis, generally in a male infant because it is X-linked. APECED declares itself through the triad of chronic mucocutaneous candidiasis, hypoparathyroidism and adrenal insufficiency, accumulating across childhood. CTLA4 and LRBA deficiency blend enteropathy, cytopenias, respiratory disease and lymphoproliferation. The thread is that the autoimmune targets are real organs and the onset is early. [4] [5]
Lymphoproliferation presents as chronic, non-tender splenomegaly and lymphadenopathy alongside autoimmune cytopenias — haemolytic anaemia, thrombocytopenia, neutropenia — that recur or persist. The child is often otherwise well, which is exactly why ALPS is mistaken for lymphoma or for Evans syndrome. The history is one of chronic, fluctuating lymph node enlargement and cytopenia over months to years, not the explosive, systemic illness of malignancy. [6] [7]
Autoinflammation presents as recurrent, unprovoked attacks with clockwork regularity. FMF brings short fevers with serositis — peritonitis, pleuritis, scrotal pain — and a migratory erysipelas-like rash. The cryopyrin-associated periodic syndromes range from cold-induced urticaria and fever (familial cold autoinflammatory syndrome) through the triad of urticaria, arthralgia and progressive sensorineural hearing loss (Muckle-Wells) to the severe neonatal-onset multisystem inflammatory disease with chronic aseptic meningitis and bony overgrowth. PFAPA, the commonest seen in general paediatrics, is a regular fever every three to eight weeks with pharyngitis, aphthous stomatitis and cervical adenitis in a child who is entirely well between attacks. [11] [13]
Differential Diagnosis
The trap with these diseases is that each mimics something commoner, so the differential is about recognising when the common explanation does not fit. [1]
For fever and organomegaly, the must-not-miss alternatives are sepsis, malignancy (lymphoma, leukaemia), and secondary HLH. A child with ALPS can look exactly like lymphoma; a child with HLH can look exactly like sepsis. The discriminating features are tempo (chronic fluctuating versus explosive progressive), wellbeing between events, and the pattern of cytopenia and acute-phase response. Empiric broad-spectrum antibiotics are mandatory until infection is excluded in any febrile, immunologically unwell child. [8] [9]
For recurrent fever, the task is to separate the common recurrent fever of childhood (serial viral infections, and PFAPA) from a hereditary recurrent fever. Family history, the regularity and duration of attacks, accompanying serositis or rash, and the acute-phase profile between attacks are the discriminators. Gattorno and colleagues' Eurofever classification criteria formalise this distinction for the hereditary recurrent fevers. [11]
For early-onset autoimmunity, the question is when to test for an underlying monogenic defect. The triggers are onset in infancy, multi-organ involvement, a family history, and resistance to standard therapy. IPEX should be considered in any male infant with enteropathy, eczema and endocrinopathy; APECED in any child with chronic mucocutaneous candidiasis and an endocrinopathy; CTLA4 or LRBA deficiency when enteropathy, cytopenias and lymphoproliferation coexist. [3] [4]
EBV-driven lymphoproliferative disease deserves a specific mention: a child with severe, recurrent or complicated EBV infection may have an underlying susceptibility such as XIAP deficiency, ITK deficiency or X-linked lymphoproliferative disease, and this enters the differential of any unusually severe EBV course. [6]
Clinical & Bedside Assessment
The first move at the bedside is always the threat gate, not the gene hunt. [8]
Ask of any child with possible immune dysregulation: is this HLH or MAS? The clinical cluster is fever plus splenomegaly plus cytopenias plus a very high ferritin, often with hepatitis, coagulopathy and neurological change. If the answer is yes or possibly yes, the assessment stops being a diagnostic exercise and becomes a resuscitation and escalation pathway — time-critical bloods, empiric treatment for suspected primary HLH, infection cover, and urgent tertiary involvement. [9] [10]
If the threat gate is clear, sort the presentation by its dominant mechanism. A focused history captures age of onset, the pattern and timing of attacks (clockwork favours PFAPA and the hereditary recurrent fevers; organ-specific autoimmunity favours dysregulation; chronic painless organomegaly favours lymphoproliferation), the organ systems involved, a careful pedigree with consanguinity, and the response to previous treatment. Steroid-responsiveness, in particular, is a useful clue: many of these diseases respond transiently to steroids, which both helps and misleads. [1] [11]
Examination then localises the mechanism. Look for the urticarial-like rash, serositis, uveitis and arthritis of autoinflammation; the organ-specific autoimmune signs (enteropathy, candidiasis, endocrinopathy) of dysregulation; and the splenomegaly and lymphadenopathy of lymphoproliferation. Plot growth and puberty, because chronic disease and chronic steroids both leave their mark, and assess development. Finally, gauge the family's understanding and the burden of a rare, potentially lifelong disease — this shapes how aggressively and how quickly you pursue the diagnosis. [7] [13]
Investigations
The cardinal error is to order a non-specific "allergy panel" or a broad autoantibody screen as the first test. The right approach is phenotype-driven, targeted testing that confirms the dominant mechanism and then seeks its genetic cause. [1]
For suspected autoinflammation, the cornerstone is the acute-phase profile during and between attacks: C-reactive protein and serum amyloid A rise markedly during an attack and normalise between them in the hereditary recurrent fevers. Serum amyloid A is also the marker for amyloidosis risk, the long-term complication of uncontrolled inflammation. A normal between-attack profile does not exclude the diagnosis, which is why documenting a typical attack biochemically matters. [12] [14]
For suspected immune dysregulation and lymphoproliferation, the first-line immunology work-up is an immunophenotype (which shows the double-negative T cells of ALPS), immunoglobulins and vaccine antibody responses (which reveal the antibody deficiency of CTLA4/LRBA), organ-specific autoantibodies directed by the phenotype, and a peripheral blood film. In ALPS, elevated vitamin B12 and soluble Fas ligand support the diagnosis alongside the characteristic immunophenotype. [6] [7]
For HLH and MAS the work-up is time-critical and runs in parallel with treatment: ferritin (markedly elevated), triglycerides (raised), fibrinogen (low), a full blood count (multilineage cytopenia), liver function and lactate dehydrogenase (hepatitis), coagulation, and a soluble interleukin-2 receptor level where available. A bone marrow aspirate showing haemophagocytosis supports the diagnosis but is neither sensitive nor specific — its main role is to exclude malignancy and confirm tissue haemophagocytosis. Natural killer cell and cytotoxic T-cell function assays and a genetic panel for familial HLH genes define the primary form. [8] [9]
Genetic testing is now central. A phenotype-guided targeted gene panel is first-line for a well-characterised case; whole-exome or whole-genome sequencing is reserved for atypical, combined or undiagnosed presentations, where it shortens the diagnostic odyssey and enables family counselling. [2]
Management — Resuscitation
The resuscitation phase exists to stop the cytokine storm and to cover infection while the diagnosis is secured. [8]
Suspected HLH or MAS is treated on clinical grounds without waiting for genetic confirmation. The HLH-2004 protocol established the principle that primary HLH is treated early with a combination of dexamethasone, etoposide and cyclosporin, with intrathecal therapy reserved for central nervous system disease, alongside supportive care for cytopenias, coagulopathy and organ failure. Jordan frames the contemporary approach similarly: treat suspected disease early, support the failing organs, control the infection trigger, and move toward haematopoietic stem cell transplant for confirmed primary disease. Empiric broad-spectrum antibiotics are essential because sepsis and HLH are clinically indistinguishable at first. [9]
For an autoinflammatory flare, acute stabilisation combines antipyretics, analgesia and, for severe attacks, a short course of corticosteroid, with early introduction of interleukin-1 blockade for the inflammasomopathies because it can abort the attack at its mechanistic root. Supportive care — fluids, transfusion for anaemia, and pain control for serositis — runs alongside. The decision between ambulatory and inpatient management rests on the severity of the flare and the risk of dehydration or organ involvement. [12] [13]
Escalation to a tertiary clinical immunology or rheumatology centre, and to paediatric intensive care for multi-organ failure, should happen early and in parallel with resuscitation rather than after it. The cytokine storm is one of the few paediatric emergencies where waiting for a firm diagnosis before treating costs lives. [8] [10]
Management — Definitive & Stepwise
Once the storm is controlled, definitive therapy is chosen to match the pathway — the mechanism that is failing — and this is where the three-way framework pays off. [2]
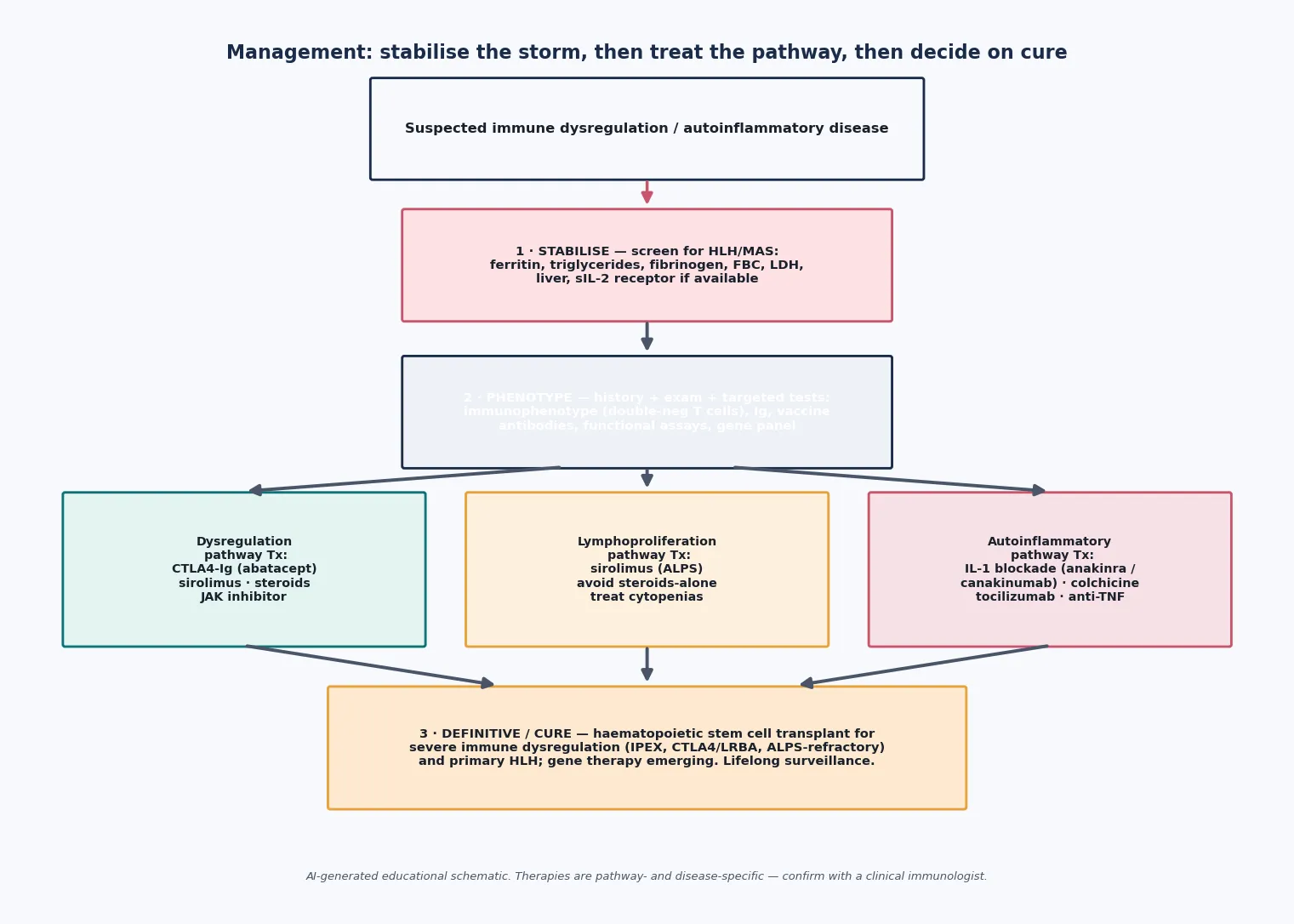
For autoinflammation, the dominant strategy is cytokine blockade. Familial Mediterranean fever is controlled lifelong with colchicine, which prevents attacks and, crucially, prevents AA amyloidosis — the Ozen EULAR recommendations make this the standard of care. The interleukin-1-mediated diseases (the cryopyrin-associated periodic syndromes, mevalonate kinase deficiency, deficiency of the interleukin-1 receptor antagonist, and increasingly TRAPS) respond to the interleukin-1 receptor antagonist anakinra or the monoclonal antibody canakinumab, which the EULAR and American College of Rheumatology points to consider endorse as first-line biological therapy. PFAPA is managed with anticipatory steroids at the start of an attack, with tonsillectomy reserved for severe, refractory cases given the syndrome's natural tendency to resolve. [12] [14]
For lymphoproliferation, the steroid-sparing mTOR inhibitor sirolimus is the first-line definitive therapy for ALPS: it controls the chronic lymphoproliferation and the autoimmune cytopenias and has displaced splenectomy, which historically caused overwhelming sepsis without curing the disease. Cytopenias are treated with the usual supportive and immunomodulatory measures, and live vaccines are avoided in splenectomised or heavily immunosuppressed children. [7]
For immune dysregulation, therapy replaces the missing brake where it can. CTLA4-Ig (abatacept) replaces the defective co-stimulatory checkpoint in CTLA4 and LRBA deficiency and can produce striking improvement. Sirolimus and steroids have a role, and sirolimus-based regimens also feature in IPEX bridging. Type-I interferonopathies and STAT1 gain-of-function disease respond to Janus kinase inhibitors (ruxolitinib, baricitinib), which damp the downstream interferon signal. [3] [17]
The definitive cure for the severe immune-dysregulation and primary-HLH diseases is haematopoietic stem cell transplant, which replaces the faulty immune system. It is indicated for primary HLH, severe IPEX, severe CTLA4 or LRBA deficiency, and refractory ALPS. Transplant does not reverse damage already done to end organs such as the islets or the kidneys, and it carries real morbidity, so it is reserved for diseases that are life-threatening or unresponsive to medical therapy. Diseases driven purely by innate cytokine excess — FMF, CAPS, and most ALPS — are managed medically for life rather than transplanted. [4] [9]
Specific Subtypes & Scenarios
The canonical diseases are best held as a small set of recognisable patterns, each with its gene, its signature and its pathway-targeted therapy. [1]
The canonical diseases, by mechanism
IPEX (FOXP3)
X-linked; male infant with enteropathy, eczema, early type-1 diabetes. Steroids and sirolimus bridge; transplant for severe disease.
APECED (AIRE)
Chronic mucocutaneous candidiasis, hypoparathyroidism, adrenal insufficiency. Replace hormones; treat candidiasis; surveillance for new components.
ALPS (FAS/FASL/CASP10)
Chronic painless splenomegaly, double-negative T cells, autoimmune cytopenias. Sirolimus first-line; avoid splenectomy.
CTLA4 / LRBA
Combined lymphoproliferation, enteropathy, cytopenias, antibody deficiency. CTLA4-Ig (abatacept) replaces the brake.
FMF (MEFV)
Short fevers with serositis and a migratory rash. Lifelong colchicine; prevents amyloidosis.
CAPS (NLRP3)
Cold-induced urticaria, Muckle-Wells triad, or neonatal multisystem inflammation. Interleukin-1 blockade, often dramatic.
PFAPA
Clockwork fevers every 3–8 weeks with pharyngitis, aphthae, adenitis; well between. Anticipatory steroids; tonsillectomy if refractory; resolves over years.
ALPS is the archetype of lymphoproliferation and a favourite of examiners. The child has chronic, non-tender splenomegaly and lymphadenopathy, a population of double-negative T cells (alpha-beta T-cell receptor positive, CD4 negative, CD8 negative), elevated immunoglobulins and vitamin B12, and autoimmune cytopenias that recur. The decisive management principle is steroid-sparing sirolimus in preference to splenectomy, which historically caused overwhelming post-splenectomy infection without durably controlling the disease. [6] [7]
IPEX is the archetype of severe immune dysregulation. The male infant presents with intractable watery or bloody diarrhoea, severe eczema, and endocrinopathy — classically type-1 diabetes or thyroiditis — and the disease is life-threatening without intervention. Immunosuppression and nutritional support bridge to definitive treatment, and haematopoietic stem cell transplant is the only curative option for severe disease. [4]
APECED is defined by its triad but is protean. Chronic mucocutaneous candidiasis is usually first, hypoparathyroidism and adrenal insufficiency follow, and ectodermal dystrophy and other autoimmune components (including autoimmune hepatitis and pneumonitis) accumulate over years. Management is hormone replacement, treatment of candidiasis, and lifelong surveillance for new endocrine and non-endocrine components. [5]
Familial Mediterranean fever is the commonest hereditary recurrent fever and the one in which treatment most clearly prevents long-term harm: lifelong colchicine controls attacks and prevents AA amyloidosis, which historically caused renal failure. The attack is a short fever with serositis — peritonitis that can mimic a surgical abdomen, pleuritis, or scrotal pain — and a migratory erysipelas-like rash on the lower legs. [14]
PFAPA is the syndrome most general paediatricians will actually see. The fevers are strikingly regular, the child is completely well between attacks, and the outlook is one of spontaneous resolution over years. Anticipatory corticosteroid at the very start of an attack aborts it in many children; tonsillectomy is reserved for severe, refractory cases because the disease is ultimately self-limiting. Stojanov and colleagues showed that PFAPA is a disorder of innate immune and T-helper-1 activation responsive to interleukin-1 blockade, which explains both the steroid response and the emerging role of interleukin-1 blockade in refractory cases. [13]
Complications & Pitfalls
The gravest pitfall is missing HLH or MAS. The cytokine storm progresses to multi-organ failure and death within days, and the features that shorten the diagnostic delay are a very high ferritin with falling cell counts in a febrile child with organomegaly — a combination that should trigger the HLH work-up immediately rather than after infection is excluded. [8] [9]
Untreated or under-treated autoinflammation leads to AA amyloidosis, the deposition of serum amyloid A in organs, principally the kidney. This is why serum amyloid A monitoring and tight control with colchicine or cytokine blockade are part of long-term care for the hereditary recurrent fevers, and why adherence matters even when the child feels well between attacks. [14]
The harms of treatment are real and bidirectional. Prolonged high-dose corticosteroids cause growth failure, infection, osteoporosis and adrenal suppression, which is why the field has moved to steroid-sparing strategies such as sirolimus for ALPS and cytokine blockade for the autoinflammatory diseases. Conversely, under-treatment from fear of immunosuppression leaves inflammation smouldering toward organ damage and amyloidosis. The balance is disease control with the least toxic effective regimen. [7] [12]
The diagnostic pitfalls are the mislabels: eczema and food allergy for IPEX; recurrent tonsillitis for PFAPA; lymphoma for ALPS; sepsis for HLH; and Crohn's disease for the enteropathy of CTLA4 or LRBA deficiency. Each costs time, and delayed genetic diagnosis harms the wider family through untreated affected siblings and missed carrier counselling. [4] [6]
Prognosis & Disposition
Prognosis in this group is determined less by the specific gene than by four things: how early the diagnosis is made, whether the correct pathway-targeted therapy is chosen, whether transplant is available when it is needed, and whether irreversible organ damage has already occurred. [2]
The outlooks differ sharply by mechanism. PFAPA resolves spontaneously over years with an excellent long-term prognosis. Familial Mediterranean fever and the cryopyrin-associated periodic syndromes are chronic but highly controllable with colchicine and interleukin-1 blockade respectively, provided adherence and surveillance are maintained. Primary HLH is transplant-dependent and carries high mortality without curative therapy. Severe IPEX and severe CTLA4 or LRBA deficiency are life-threatening without transplant. ALPS is generally controllable with sirolimus and carries a small long-term lymphoma risk that warrants surveillance. [9] [14]
Urgent tertiary referral is driven by the threat gate and by severity: any suspected HLH or MAS, ALPS with severe or refractory cytopenias, suspected IPEX, and CTLA4 or LRBA deficiency all need a clinical immunologist and usually a transplant centre. Less acute presentations — a stable child with PFAPA or controlled FMF — can be shared between primary care, general paediatrics and a specialist service. [8]
Because these are lifelong diseases, the transition from paediatric to adult immunology or rheumatology care must be structured and planned, with a written flare action plan, genetic counselling for the patient and family, school and childcare education, and an individualised immunisation plan that accounts for live vaccines and immunosuppression. [4]
Special Populations
Age of onset changes the differential. Neonatal and infantile presentations point toward IPEX, severe combined immune dysregulation, neonatal-onset CAPS and familial HLH. The toddler and preschool years are the classic window for PFAPA and for the declaration of FMF and mevalonate kinase deficiency. School-age children declare systemic juvenile idiopathic arthritis and its MAS risk, chronic recurrent multifocal osteomyelitis, Blau syndrome and TRAPS. Adolescents face transition, adherence to lifelong biologics, and reproductive and genetic counselling. [11] [15]
Consanguineous families and communities with founder mutations face a different detection and counselling pathway: autosomal recessive diseases cluster, carrier testing and prenatal or preimplantation options become central, and culturally meaningful framing through a trained interpreter is essential for Indigenous, refugee and migrant families managing a rare genetic diagnosis. [4]
Children with disability, neurodiversity or feeding difficulties face particular challenges with chronic immune therapy — adherence to biologics, the burden of frequent blood monitoring, and the interaction between immunosuppression and their other care needs. Socioeconomic disadvantage affects access to expensive biologics, transplant and the long-term surveillance these diseases require, and the clinician's role includes advocating for subsidised access and coordinated care. [7]
Evidence, Guidelines & Regional Differences
The framework on this page rests on the IUIS classification of inborn errors of immunity — the foundational nine-group structure from Picard and the 2015 committee, expanded in the 2022 update by Tangye and colleagues — which formally groups these diseases by mechanism rather than by the organ they happen to attack. [1] [2]
For autoinflammatory disease, the evidence base is now guideline-graded. Gattorno and the Eurofever and PRINTO groups produced classification criteria for the autoinflammatory recurrent fevers, and Romano and colleagues' 2021 EULAR and American College of Rheumatology points to consider set out diagnosis, management and monitoring for the interleukin-1-mediated diseases — the cryopyrin-associated periodic syndromes, TRAPS, mevalonate kinase deficiency and deficiency of the interleukin-1 receptor antagonist. For familial Mediterranean fever, the Ozen EULAR recommendations establish lifelong colchicine as the standard of care. [11] [12] [14]
For haemophagocytic lymphohistiocytosis, the Henter HLH-2004 guidelines remain the standard diagnostic and treatment framework, and Jordan's treatment review frames the contemporary, transplant-directed approach. The field continues to debate the diagnostic thresholds — how high a ferritin, how reliable a soluble interleukin-2 receptor level — but not the principle of treating suspected disease early. [8] [9]
ANZ: specialist paediatric immunology and rheumatology are concentrated in tertiary children's hospitals; rare-disease registries support care; subsidised access to biologics varies by region and usually requires specialist authorisation; culturally safe care for Aboriginal, Torres Strait Islander, Maori and Pacific peoples and migrant families is mandated. UK: NHS highly specialised services and national commissioning cover primary immunodeficiency and autoinflammatory disease; RCPCH and BSACI pathways apply; robust gene-panel and whole-genome sequencing run through the Genomic Medicine Service. North America: the US Immune Deficiency Foundation network, the US and Canadian Immunodeficiency Network, and the CARRA consortium support care; access to biologics and transplant is insurance-driven. Europe: the EULAR, PRINTO and Eurofever collaboration drives classification criteria and care standards through shared rare-disease registries.
[12]The controversies live at the boundaries: where autoimmunity ends and autoinflammation begins in combined-feature patients such as those with CTLA4 or LRBA deficiency; whether to use early whole-exome sequencing or a phenotype-guided gene panel first; and how emerging cellular therapies — engineered regulatory T cells for IPEX, gene therapy, and refined JAK-inhibitor use — will change the landscape over the next decade. [3] [17]
Exam Pearls
Hold the three mechanisms and their canonical genes as a single sentence, because the examiner will ask for it: tolerance failure (FOXP3, AIRE, CTLA4, LRBA), apoptosis failure (FAS, FASL, CASP10), and innate cytokine over-activation (NLRP3, MEFV, TNFRSF1A, MVK). [1]
The HLH or MAS red-flag cluster is exam gold: fever plus splenomegaly plus cytopenias plus a very high ferritin, and the HLH-2004 principle of early treatment with dexamethasone, etoposide and cyclosporin moving to transplant. [8]
The APECED triad — chronic mucocutaneous candidiasis, hypoparathyroidism, adrenal insufficiency, with AIRE — is a classic recall item. The ALPS hallmark — double-negative T cells (alpha-beta T-cell receptor positive, CD4 and CD8 negative) with chronic splenomegaly and autoimmune cytopenias, treated with steroid-sparing sirolimus — is its lymphoproliferation counterpart. [5] [6]
Know which diseases are transplant-curable — primary HLH, severe IPEX, severe CTLA4 or LRBA deficiency — versus those managed medically for life — FMF, CAPS, and most ALPS. And know the IL-1-dominant hereditary recurrent fevers (CAPS, FMF, mevalonate kinase deficiency, deficiency of the interleukin-1 receptor antagonist) for the dramatic response to anakinra or canakinumab that the EULAR and American College of Rheumatology points to consider endorse. [9] [12]
References
- [1]Picard C Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. Journal of Clinical Immunology, 2015.PMID 26482257
- [2]Tangye SG Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology, 2022.PMID 35748970
- [3]Kuehn HS Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science, 2014.PMID 25213377
- [4]Verbsky JW Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Current Opinion in Pediatrics, 2013.PMID 24240290
- [5]Ferré EMN Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Frontiers in Pediatrics, 2021.PMID 34790633
- [6]Rieux-Laucat F Scaling the tips of the ALPS. Biomedical Journal, 2021.PMID 34438083
- [7]George LA Optimal Management of Autoimmune Lymphoproliferative Syndrome in Children. Paediatric Drugs, 2016.PMID 27139496
- [8]Henter JI HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood and Cancer, 2007.PMID 16937360
- [9]Jordan MB How I treat hemophagocytic lymphohistiocytosis. Blood, 2011.PMID 21828139
- [10]Ravelli A Macrophage Activation Syndrome. Hematology/Oncology Clinics of North America, 2015.PMID 26461152
- [11]Gattorno M Classification criteria for autoinflammatory recurrent fevers. Annals of the Rheumatic Diseases, 2019.PMID 31018962
- [12]Romano M The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Annals of the Rheumatic Diseases, 2022.PMID 35623638
- [13]Stojanov S Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proceedings of the National Academy of Sciences, 2011.PMID 21478439
- [14]Ozen S EULAR recommendations for the management of familial Mediterranean fever. Annals of the Rheumatic Diseases, 2016.PMID 26802180
- [15]Wouters CH Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatric Rheumatology, 2014.PMID 25136265
- [16]Holland SM STAT3 mutations in the hyper-IgE syndrome. New England Journal of Medicine, 2007.PMID 17881745
- [17]Toubiana J Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood, 2016.PMID 27114460