Paeds · allergy-and-immunology
Phagocyte disorders
Also known as Chronic granulomatous disease · CGD · Leukocyte adhesion deficiency · LAD · Severe congenital neutropenia · Chédiak-Higashi syndrome · Congenital neutropenia · Innate immune deficiency
Fellowship topic on phagocyte disorders — the primary immunodeficiencies in which neutrophils and monocytes fail to kill microbes, recurrent tissue invasive infection drives the presentation, and inflammation paradoxically coexists with infection. Chronic granulomatous disease (CGD) is the prototype and the most tested: a defective NADPH oxidase prevents the respiratory burst, so catalase-positive organisms (Staphylococcus aureus, Burkholderia cepacia complex, Serratia, Nocardia, Aspergillus) survive inside phagocytes and the host responds with granulomatous inflammation that obstructs hollow viscera. X-linked gp91phox (CYBB) is the commonest genotype, autosomal recessive p47phox (NCF1) the next, and the dihydrorhodamine (DHR) flow cytometry assay is the diagnostic test. Leukocyte adhesion deficiency (LAD) presents with delayed cord separation, pus-less infection, and striking leukocytosis; severe congenital neutropenia with overwhelming bacterial sepsis in early infancy and a long-term myelodysplasia risk; Chédiak-Higashi syndrome with giant intracellular granules, partial albinism, and a fatal accelerated phase. The curative therapy for the severe forms is haematopoietic stem cell transplantation, and antimicrobial prophylaxis (co-trimoxazole and an anti-mould antifungal) is the backbone of medical management. The page owns CGD, LAD-1/2/3, congenital and cyclic neutropenia, Chédiak-Higashi, specific granule deficiency, and Shwachman-Diamond, and cross-links the combined immunodeficiency leaf for SCID and the antibody-deficiency leaf for humoral defects.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The CGD organism cluster and the rule of thumb
Overview & Definition
Picture a three-year-old boy admitted for the third time in a year — once for a staphylococcal liver abscess that needed weeks of intravenous antibiotics, once for a necrotising pneumonia that turned out to be caused by Serratia marcescens, and now for a swollen, chronically inflamed gum that a biopsy calls granulomatous. His neutrophil count is normal, and each time the infection has produced abundant pus, yet the organisms keep recurring. The clue is the organism list itself — Staphylococcus, Serratia, and a granulomatous response — and it points straight to a phagocyte killing defect. This child has chronic granulomatous disease, and the single test that confirms it, the dihydrorhodamine flow cytometry assay, measures the oxidative burst his neutrophils cannot generate. [4] [5]
The phagocyte disorders are the primary immunodeficiencies in which neutrophils and monocytes — the innate effector cells that migrate into infected tissue, ingest microbes, and kill them with reactive oxygen species and granule enzymes — fail at one of these steps. The 2024 IUIS classification, led by Poli and colleagues, groups them under congenital defects of phagocyte number or function, and the field divides cleanly along three mechanistic axes: a failure of killing (CGD, the largest group), a failure of adhesion and migration (leukocyte adhesion deficiency), and a failure of production or granule packaging (severe congenital neutropenia, cyclic neutropenia, Chédiak-Higashi, specific granule deficiency). Each axis produces a recognisable clinical phenotype, and recognising the phenotype is how the diagnosis is made at the bedside. [1] [2]
What unifies these disorders, and what the examination tests, is that the recurrent infection they cause is driven by a predictable cluster of organisms and by a paradoxical tissue response. In CGD the catalase-positive bacteria and Aspergillus survive because the respiratory burst fails; the host responds with granulomatous inflammation that can obstruct the gut or urinary tract. In LAD the neutrophils cannot migrate, so infection smoulders without pus while the neutrophil count climbs. In congenital neutropenia the marrow simply cannot make enough neutrophils, and the child presents with overwhelming bacterial sepsis. Understanding why each disease produces its particular fingerprint is the key to recognising all of them. [3] [4]
The clinical task has two halves. The first is recognition and protection — suspect a phagocyte disorder from the organism list or the bedside paradox, confirm with the DHR assay or flow cytometry, and start prophylaxis and infection surveillance. The second is the curative decision — haematopoietic stem cell transplantation for the severe forms, particularly CGD and LAD, in which the transplant replaces the defective phagocyte lineage with healthy donor cells. The 712-patient multicentre cohort reported by Chiesa and colleagues established that transplant works across genotypes and ages, making the decision not whether to transplant but when and with what donor. A candidate who diagnoses CGD but does not know the organism cluster, the DHR assay, or the transplant evidence fails the question. [4] [7]
Classification
Classification of the phagocyte disorders matters because it directs the diagnostic test, predicts the inheritance and the genetic counselling, and informs the transplant decision. The most useful clinical axis is the mechanistic failure — killing, adhesion, or production — because each produces a distinctive clinical fingerprint and points to a specific confirmatory investigation. [1] [3]
Chronic granulomatous disease is the prototype killing defect and the commonest clinically significant phagocyte disorder. The X-linked form, from mutations in CYBB encoding the gp91phox subunit of the NADPH oxidase, accounts for roughly two-thirds of cases and affects boys; the autosomal recessive forms, most often p47phox (NCF1, about a quarter of all CGD) but also p67phox (NCF2), p22phox (CYBA), and p40phox (NCF4), together account for the remainder and affect both sexes. The Kuhns cohort, which combined comprehensive genetic and flow cytometric analysis, established that p47phox deficiency arises almost always from recombination between the NCF1 gene and its two pseudogenes — a recurrent mutational mechanism the candidate should be able to name. [4] [6]
The adhesion defects are leukocyte adhesion deficiency types 1, 2, and 3. LAD-1, from ITGB2 encoding the β2 integrin CD18, is the classic and commonest form and the one the examination tests; LAD-2, from SLC35C1 causing a generalised fucose transporter defect, adds developmental delay and the Bombay blood group; LAD-3, from FERMT3 encoding kindlin-3, adds a bleeding tendency from platelet dysfunction because kindlin-3 is shared between leucocyte and platelet integrin activation. The Hanna and Etzioni review set out the three-type framework that remains the working classification. [9] [8]
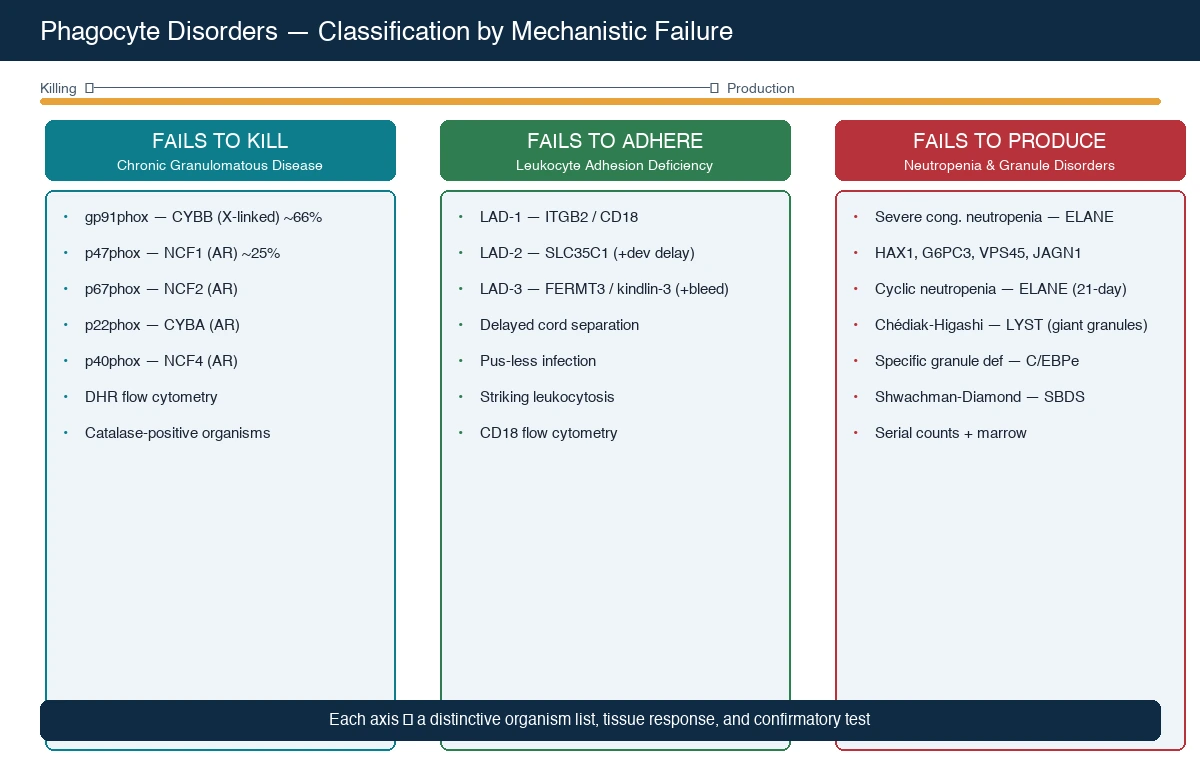
The production defects are severe congenital neutropenia and cyclic neutropenia. Severe congenital neutropenia is genetically heterogeneous — ELANE (the commonest, autosomal dominant) is joined by HAX1 (autosomal recessive, with an associated neurological phenotype), G6PC3, VPS45, and JAGN1 — but all share a profound, persistent neutropenia with early severe bacterial infection and a long-term risk of myelodysplasia and acute myeloid leukaemia. Cyclic neutropenia, also from ELANE, runs a 21-day cycle of neutrophil nadir and is generally milder. The Skokowa primer in Nature Reviews Disease Primers is the comprehensive reference for the congenital neutropenias. [10]
The granule-packaging defects are Chédiak-Higashi syndrome, from LYST, and specific granule deficiency, from C/EBPe. Chédiak-Higashi produces giant intracytoplasmic granules in all granule-bearing cells, partial oculocutaneous albinism, a mild bleeding tendency from platelet-dense-granule deficiency, recurrent pyogenic infection, and a frequently fatal accelerated (haemophagocytic lymphohistiocytic) phase. Specific granule deficiency is rare and produces atypical infection from the absence of the neutrophil's defence granule contents. Shwachman-Diamond syndrome, from SBDS, combines neutropenia with pancreatic exocrine insufficiency and skeletal changes, and is grouped here because neutropenia is its dominant haematological feature. [3] [11]
Epidemiology & Risk Factors
Where do the phagocyte disorders sit in paediatric practice, and who is at risk? The incidence of CGD is approximately one in two hundred thousand live births, and because the commonest form is X-linked, the majority of affected children are boys. Consanguinity increases the rate of the autosomal recessive forms — p47phox, p67phox, p22phox — which is relevant in populations with high rates of cousin marriage and in some migrant and refugee communities. A family history of male relatives who died in infancy or childhood from deep-seated infection, or of male relatives with a known CGD diagnosis, is the single most important historical clue for X-linked CGD and should trigger maternal carrier testing. [4] [5]
LAD-1 is rare, with an estimated incidence of about one in a million, but it is the form the examination tests because its neonatal presentation — delayed cord separation with omphalitis and a striking leukocytosis — is unforgettable. Severe congenital neutropenia has an estimated incidence of roughly two to eight per million, with ELANE mutations accounting for about half of cases. Chédiak-Higashi syndrome is rarer still, with only a few hundred cases reported worldwide, but its clinical triad of albinism, giant granules, and recurrent infection makes it a high-yield examination topic. [8] [10]
The cofactors that worsen outcome are preventable and the candidate must name them. Administration of the live BCG vaccine to a child with an undiagnosed severe phagocyte disorder — particularly CGD and LAD — can cause local or disseminated BCG disease, because the attenuated mycobacterium, which a healthy phagocyte contains, persists in a defective one. Delayed diagnosis of congenital neutropenia is dangerous because the first severe infection can be fatal before the marrow failure is recognised, and missed bone-marrow surveillance in congenital neutropenia allows myelodysplasia or leukaemia to declare late. The accelerated phase of Chédiak-Higashi is the chief cause of death in that syndrome, and a fever with hepatosplenomegaly in a child with Chédiak-Higashi is the presentation that demands emergency haematology involvement. [11] [12]
Pathophysiology
To understand why CGD is so distinctive, you have to understand the respiratory burst — because the whole disease is a lesson in what happens when the phagocyte cannot convert oxygen into a microbe-killing weapon. When a neutrophil ingests a bacterium or fungus, the NADPH oxidase enzyme complex assembles on the phagolysosomal membrane and pumps electrons from NADPH onto molecular oxygen, generating superoxide anion, which dismutates to hydrogen peroxide. Myeloperoxidase, stored in the neutrophil's azurophilic granules, then uses that hydrogen peroxide plus a halide ion to generate hypochlorous acid — bleach — the final microbe-killing agent. The whole cascade is the respiratory burst, and without it the phagocyte cannot kill catalase-positive organisms. [4] [5]
The reason the CGD organism cluster is so specific lies in a single enzymatic fact. Myeloperoxidase needs hydrogen peroxide as its fuel, and most of that peroxide comes from the phagocyte's own NADPH oxidase. Catalase-positive organisms — Staphylococcus aureus, Burkholderia, Serratia, Nocardia, Aspergillus — carry the enzyme catalase, which rapidly breaks down any peroxide the microbe itself makes. A normal neutrophil does not care, because it makes its own peroxide via the respiratory burst. A CGD neutrophil makes no peroxide, and the catalase-positive organism destroys the little it produces, so the microbe survives inside the phagocyte. Catalase-negative organisms such as Streptococcus pneumoniae and the streptococci do not trouble the CGD patient, because they cannot destroy their own peroxide — that peroxide diffuses into the phagolysosome and fuels myeloperoxidase anyway. This is why CGD patients get staphylococcal and Aspergillus infection but not streptococcal infection, and it is the single most elegant mechanistic point in paediatric immunology. [4] [5]
The NADPH oxidase is a multi-subunit enzyme, and the genetics map onto its components. The membrane-bound cytochrome b558 is composed of gp91phox (encoded by CYBB on the X chromosome) and p22phox (CYBA, autosomal). Upon activation, the cytosolic subunits p47phox (NCF1), p67phox (NCF2), and p40phox (NCF4) translocate to the membrane and assemble the active oxidase. A mutation in any one subunit abolishes the burst, but the commonest is X-linked gp91phox, followed by autosomal recessive p47phox. The Kuhns study, combining comprehensive genetic and flow cytometric analysis across a large cohort, confirmed that p47phox deficiency arises almost exclusively from recombination between NCF1 and its two flanking pseudogenes — a gene-conversion event that deletes the GT dinucleotide start of exon 2 — and established the modern molecular diagnostics of the disease. [6]
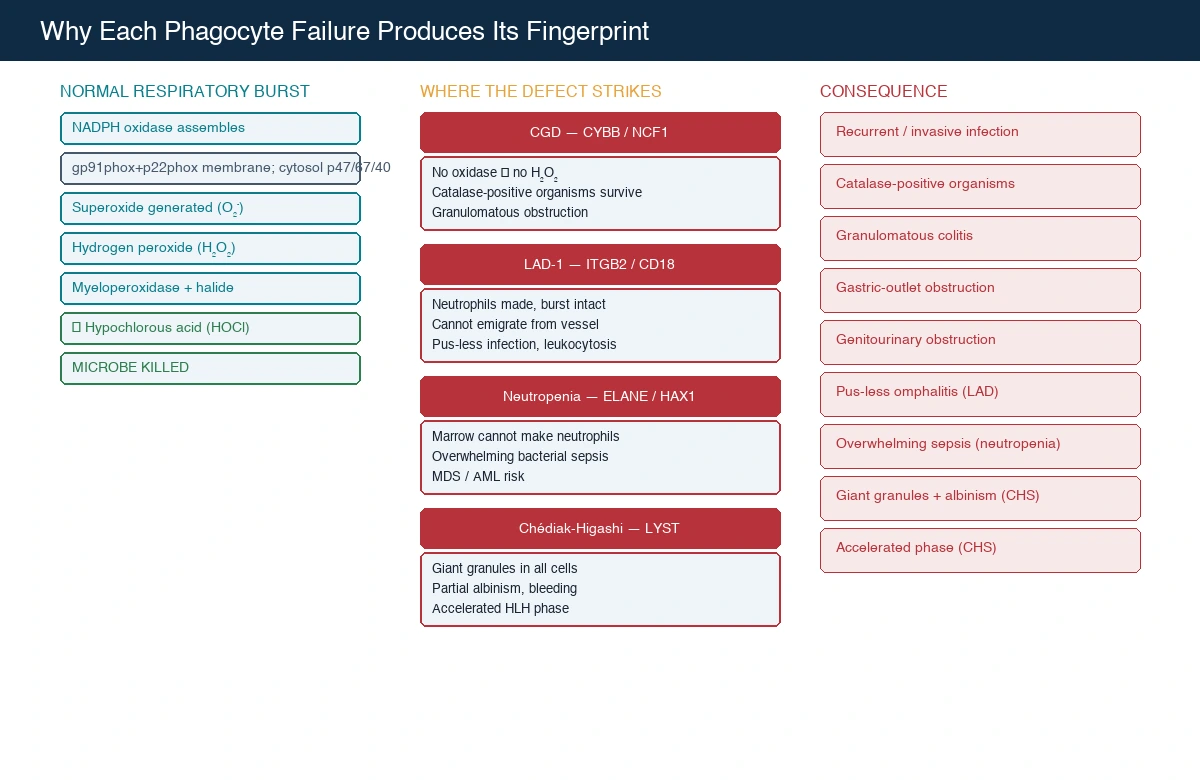
The inflammation in CGD is the other half of the pathophysiology, and it is what produces the granulomatous complications that dominate the long-term course. Because the neutrophil cannot kill the organism, macrophages accumulate at the site and wall off the surviving microbes with granulomatous inflammation — a response that is appropriate for containing infection but, in CGD, becomes dysregulated. Granulomas obstruct the gastrointestinal tract (a Crohn-like colitis and gastric-outlet obstruction), the genitourinary tract (recurrent hydronephrosis and bladder obstruction), and the hepatobiliary tree. This is why the CGD patient has both infection and inflammation — the dual phenotype that the candidate must explain — and why anti-inflammatory therapy, including corticosteroids, is sometimes needed alongside antimicrobials to control the obstructive granulomas. [4] [5]
Leukocyte adhesion deficiency works by a completely different mechanism. Neutrophils must adhere to the vascular endothelium at the site of infection before they can emigrate into the tissue, and this adhesion depends on the β2 integrin family — CD11a/CD18, CD11b/CD18, CD11c/CD18 — whose shared β2 subunit (CD18) is encoded by ITGB2. In LAD-1, absent or dysfunctional CD18 abolishes the integrin-mediated tight adhesion step, so neutrophils cannot leave the bloodstream. They are manufactured in normal numbers and their oxidative burst is intact, but they never reach the infection, so the site smoulders without pus while the neutrophil count climbs to extraordinary levels. The van de Vijver review set out the molecular basis of all three LAD types. [8] [9]
Severe congenital neutropenia is a failure of production. The dominant form, from ELANE, produces a misfolded neutrophil elastase protein that triggers apoptosis of myeloid precursors in the bone marrow — an unfolded-protein response that halts granulopoiesis. HAX1 deficiency is autosomal recessive and adds a neurological phenotype because HAX1 has a role in neuronal survival. Whatever the gene, the result is a profound, persistent neutropenia — an absolute neutrophil count below 0.5 × 10⁹/L — and the long-term risk of myelodysplasia and leukaemia, which the Skokowa primer attributes to the genomic instability and clonal evolution that accompany chronic granulopoietic stress and, in some genotypes, G-CSF exposure. [10]
Chédiak-Higashi syndrome is a granule-trafficking defect. The LYST gene product regulates lysosomal and granule biogenesis, and its loss produces abnormally large granules that fuse inappropriately in all granule-bearing cells — neutrophils (giant azurophilic granules that impair chemotaxis and degranulation), melanocytes (the partial albinism, because melanin cannot be distributed), platelets (a bleeding tendency from dense-granule deficiency), and natural killer cells (impaired cytotoxicity). The accelerated phase is a haemophagocytic lymphohistiocytic syndrome triggered by an infection, usually Epstein-Barr virus, in which the impaired cytotoxic function cannot control lymphocyte proliferation and macrophage activation runs unchecked. [11]
Clinical Presentation
The CGD child presents, almost always, in the first few years of life with recurrent or persistent deep-seated infection by the characteristic organism cluster. The infections are unusual in their depth and their causative organisms — a liver abscess from Staphylococcus aureus, a necrotising pneumonia from Burkholderia or Serratia, a brain or cutaneous abscess from Nocardia, an invasive pulmonary or disseminated infection from Aspergillus, or osteomyelitis from Serratia. Suppuration is abundant — unlike LAD, CGD neutrophils arrive in normal numbers — yet the infections fail to clear with standard courses and recur. The pattern of a normal or high neutrophil count with recurrent abscess-forming infection by the CGD cluster is the clinical fingerprint. [4] [5]
The inflammatory granulomatous complications are equally characteristic and often bring the child to specialist attention. A Crohn-like colitis with diarrhoea, abdominal pain, and weight loss is easily mistaken for inflammatory bowel disease until the infectious history and the organism cluster prompt the DHR assay. Gastric-outlet obstruction from antral granulomas causes vomiting and failure to thrive. Genitourinary granulomatous obstruction causes recurrent hydronephrosis and bladder-outlet symptoms. Perianal abscesses and fistulae, oral ulceration, and a characteristic swollen, inflamed, granulomatous gingivitis complete the picture. The candidate who hears recurrent abscesses plus granulomatous obstruction thinks CGD. [4] [5]
LAD-1 presents in the neonatal period with a triad the candidate must know by heart: delayed separation of the umbilical cord beyond three weeks, omphalitis of the cord stump with a surprising absence of pus, and a markedly elevated neutrophil count. Severe periodontitis with loss of teeth, recurrent skin infection that heals poorly and leaves dysplastic (cigarette-paper) scars, and delayed wound healing follow. The moderate phenotype presents later with less severe periodontal disease and recurrent skin infection. LAD-2 adds developmental delay, the Bombay blood group, and the rare fucose-related features. LAD-3 adds a bleeding tendency from platelet integrin dysfunction, which may be the presenting feature. [8] [9]
Severe congenital neutropenia presents in the first months of life with overwhelming bacterial sepsis, omphalitis, skin and soft-tissue infection, and sometimes deep abscesses — driven by the profound neutropenia. Oral ulceration and gingivitis are common, and chronic mouth infection may be the first sign in milder cases. Cyclic neutropenia presents with a recurring 21-day cycle of fever, mouth ulceration, and skin or throat infection that tracks the neutrophil nadir, and the cycle is the diagnostic clue. Chédiak-Higashi presents with recurrent pyogenic infection (especially Staphylococcus), partial oculocutaneous albinism that is often subtle, a mild bleeding tendency, and progressive neurological involvement — and, at any point, the accelerated phase with fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, and haemophagocytosis. [10] [11]
Differential Diagnosis
The differential diagnosis of recurrent invasive infection with a normal neutrophil count is the differential of CGD, and the first distinction is secondary immunodeficiency. Children on immunosuppressive therapy, with poorly controlled diabetes, with severe malnutrition, or with secondary neutropenia from chemotherapy can present with deep-seated infection that mimics a primary phagocyte defect. The history distinguishes these, but the clinician must order the DHR assay if the organism cluster fits, because CGD is missed when it is attributed to bad luck or to a secondary cause. [4] [5]
Within the primary immunodeficiencies, the key distinction is between the phagocyte defects and the combined or antibody defects. Severe combined immunodeficiency presents earlier, in the first months of life, with opportunistic infection (Pneumocystis, cytomegalovirus), lymphopenia, and failure to thrive — a different organism cluster and a different cell count. The antibody deficiencies present later with recurrent sinopulmonary bacterial infection and normal neutrophil counts. The phagocyte disorders, by contrast, present with the CGD organism cluster or with the bedside paradox of inflammation without pus (LAD), and the confirmatory tests — DHR for CGD, flow cytometry for LAD — are specific to the innate effector limb. [3] [4]
The granulomatous colitis of CGD must be distinguished from Crohn disease, and this is a frequent examination trap. Both produce transmural granulomatous inflammation of the colon, but the CGD colitis accompanies a history of recurrent abscess-forming infection, often has perianal involvement, and shows pigmented macrophages on biopsy. The DHR assay settles the question. Equally, the LAD neonate with omphalitis and leukocytosis must be distinguished from a simple umbilical infection — the delayed cord separation and the absent pus are the clues — and from leukaemia, which also produces a high neutrophil count, distinguished by the blast cells on the blood film and the bone marrow. [4] [8]
Severe congenital neutropenia must be distinguished from benign ethnic neutropenia — a lower neutrophil count that is normal for some populations, with no infection risk — and from transient neutropenia of viral infection, autoimmune neutropenia of infancy (which is benign and self-limiting), and cyclic neutropenia (which the serial counts reveal). Chédiak-Higashi is distinguished from other causes of partial albinism and recurrent infection by the giant granules on the blood film, which are pathognomonic, and from haemophagocytic lymphohistiocytosis by the preceding history of albinism and granule abnormality. [10] [11]
Clinical & Bedside Assessment
Assess the child with a suspected phagocyte disorder by looking at the whole infection history, because the pattern is the diagnosis. Document every infection and its causative organism — the organism list is more informative than any single feature, and the CGD cluster should be actively sought. Examine the skin for abscesses, the poor dysplastic scarring of LAD, and the partial albinism of Chédiak-Higashi. Examine the mouth for the granulomatous gingivitis of CGD, the severe periodontitis of LAD, and the oral ulceration of neutropenia. Palpate the abdomen for hepatosplenomegaly, which in Chédiak-Higashi heralds the accelerated phase. Examine the umbilicus of any neonate with infection — a cord still attached beyond three weeks with omphalitis is LAD until CD18 is checked. [4] [8]
For the CGD child, assess the inflammatory complications systematically. Ask about abdominal pain, vomiting, and diarrhoea that suggest the granulomatous colitis or gastric-outlet obstruction. Ask about urinary symptoms and check for hydronephrosis on imaging, because genitourinary granulomatous obstruction is common and may be asymptomatic until renal function is threatened. Inspect the perianal region for fistulae and abscesses. Plot the growth chart, because the combination of chronic infection and inflammatory bowel disease often causes failure to thrive. The assessment is of a multisystem disease in which infection and inflammation coexist and each must be tracked. [4] [5]
The anchoring history is as important as the examination. Ask about the age of onset and the specific organisms, because the CGD cluster points straight to the diagnosis. Ask about the vaccine history in detail — did the child receive neonatal BCG, and was there any local or disseminated reaction? Ask about family history, specifically male relatives with recurrent infection or early death (X-linked CGD) and consanguinity (autosomal recessive forms). For the neutropenic child, ask about the cycle of symptoms that suggests cyclic neutropenia, and about any cytogenetic or bone-marrow results that suggest evolving myelodysplasia. Document every hospitalisation and every organism isolated. [6] [10]
For the Chédiak-Higashi child, the assessment includes the neurological examination, because progressive cerebellar and peripheral nerve involvement develops and may be the dominant long-term problem. Assess the hair and skin tone for the partial albinism, which may be subtle in fair-skinned families but is usually evident in comparison with parents and siblings. Check the blood film for the giant granules that are pathognomonic, and maintain a low threshold for urgent assessment whenever fever and hepatosplenomegaly appear, because the accelerated phase is the chief cause of death and demands immediate haematology involvement. [11]
Investigations
The first investigation in any suspected phagocyte disorder is the full blood count with the differential, because the neutrophil count itself is diagnostic for the neutropenias and a powerful clue for the others. Profound neutropenia (below 0.5 × 10⁹/L) persistent from early infancy is severe congenital neutropenia; a 21-day cyclic pattern on serial counts is cyclic neutropenia. A normal or high neutrophil count with recurrent invasive infection points to CGD, and an extraordinarily high neutrophil count with pus-less infection points to LAD. The blood film adds the giant granules of Chédiak-Higashi and the blast cells of leukaemia. [4] [10]
The decisive investigation for CGD is the dihydrorhodamine (DHR) 123 flow cytometry assay. Dihydrorhodamine is a dye that fluoresces when oxidised; in a normal neutrophil, the respiratory burst oxidises it after stimulation with phorbol myristate acetate, producing a bright fluorescence the flow cytometer quantifies. In X-linked CGD the fluorescence is virtually absent; in the autosomal recessive p47phox form it is reduced but detectable, a mosaic-like pattern that reflects residual oxidase activity in a subpopulation. The DHR assay has replaced the older nitroblue tetrazolium (NBT) slide test because it is faster, quantitative, and works on the flow platform — and because it distinguishes the X-linked from the autosomal recessive pattern. Genetic testing (a targeted gene panel covering CYBB, NCF1, NCF2, CYBA, and NCF4) then confirms the molecular diagnosis, informs the carrier testing for X-linked forms, and guides the transplant decision. [4] [6]
For LAD, the decisive investigation is flow cytometry for the β2 integrin CD18 (and its partner CD11b), which shows absent or severely reduced expression in LAD-1. For Chédiak-Higashi, the blood film shows the pathognomonic giant granules and genetic testing confirms the LYST mutation. For the congenital neutropenias, serial neutrophil counts establish the pattern, a bone-marrow aspirate and biopsy assess the myeloid maturation and exclude marrow failure, and serial marrow cytogenetics monitor for the clonal evolution (monosomy 7, acquired trisomy 21) that heralds myelodysplasia or leukaemia. Immunoglobulin levels are usually normal in the phagocyte disorders, which is itself a useful negative finding that distinguishes them from the antibody deficiencies. [8] [11]
Active infection must be investigated thoroughly and aggressively because the infectious burden determines the transplant risk and the urgency of definitive therapy. Request imaging of the likely site — ultrasound or CT for liver abscess, high-resolution CT for invasive pulmonary aspergillosis, MRI for cerebral lesions — and obtain tissue wherever possible for culture and histology, because the organism identification both guides therapy and, in the case of the CGD cluster, points to the diagnosis. Inflammatory markers and the inflammatory complications (colitis, obstruction) are assessed with the appropriate endoscopy and imaging, and the candidate should be able to articulate that infection control and inflammation control are parallel management streams in CGD. [4] [7]
Management — Resuscitation
When a phagocyte disorder is suspected or confirmed, the first priority is to control active infection aggressively and to prevent the iatrogenic harms that worsen outcome. The child with an invasive CGD infection — a staphylococcal liver abscess, an invasive Aspergillus pneumonia, a Nocardia brain abscess — needs prolonged, organism-targeted intravenous therapy guided by culture and susceptibility, because short courses fail and recurrence is the rule. Surgical drainage of abscesses is often required, because the killed-but-not-cleared microbial burden in a CGD neutrophil perpetuates the granulomatous response. [4] [5]
Stop the live BCG vaccine and withhold other live vaccines while a severe phagocyte disorder is being investigated or confirmed. The live BCG vaccine, safe in an immunocompetent child, can cause local or disseminated disease in CGD and LAD because the attenuated mycobacterium, which a healthy phagocyte contains, persists in a defective one. The recommendations of the Medical Advisory Committee of the Immune Deficiency Foundation, published by Shearer and colleagues, set out the vaccine approach for immunodeficient patients: live bacterial vaccines are withheld from patients with severe phagocyte defects, and the rotavirus vaccine is withheld from infants with severe combined immunodeficiency but is generally acceptable in phagocyte disorders. The BCG caution is the one to remember for this leaf. [12]
Start antimicrobial prophylaxis immediately on diagnosis of CGD. Co-trimoxazole provides continuous prophylaxis against the catalase-positive bacterial cluster — particularly Staphylococcus and Nocardia — and itraconazole or posaconazole provides anti-mould prophylaxis against Aspergillus. The prophylaxis regimen is the single most important determinant of infection-free survival in the medical management of CGD, and the Arnold and Heimall review established it as the standard of care. Interferon-gamma, once widely used as immunomodulatory prophylaxis, is now used less routinely in regions where mould-active antifungal prophylaxis is standard, but remains a consideration in some centres. [5] [7]
Co-trimoxazole (prophylaxis in CGD)
Dose
5 mg/kg/day of the trimethoprim component
For the neutropenic child, the acute management of a febrile episode is the febrile-neutropenia protocol — prompt broad-spectrum intravenous antibiotics after blood cultures, because the neutropenic child cannot localise infection and a minor fever can herald overwhelming sepsis. G-CSF (granulocyte colony-stimulating factor) is the mainstay of long-term management for severe congenital neutropenia, raising the neutrophil count and reducing infection frequency, though it does not abolish the myelodysplasia risk. For the Chédiak-Higashi child in the accelerated phase, the management is urgent haematology-led treatment of the haemophagocytic syndrome — the HLH-2004 protocol of chemotherapy and immunosuppression as a bridge to curative transplant. [10] [11]
Management — Definitive & Stepwise
The curative therapy for the severe phagocyte disorders — CGD and LAD and Chédiak-Higashi — is haematopoietic stem cell transplantation, which replaces the defective phagocyte lineage with healthy donor cells. The principle is the same as in severe combined immunodeficiency: a normal myeloid system grows back from the donor stem cells, and the new neutrophils generate a normal respiratory burst (CGD), express normal CD18 (LAD), and carry normal granules (Chédiak-Higashi). Unlike SCID, CGD transplant typically requires conditioning to create marrow space, because the CGD bone marrow is cellular rather than empty. [4] [7]
The evidence for CGD transplant is anchored by the multicentre cohort reported by Chiesa and colleagues, which analysed 712 children and adults transplanted for CGD across European and international centres. The study confirmed overall survival of roughly 70 to 95 percent depending on age, genotype, infection status, and donor, with matched sibling donors giving the best outcomes and the best results achieved when transplant occurs before severe infection-driven organ damage. The donor hierarchy is the same as for SCID — matched sibling, matched unrelated, haploidentical — and the conditioning is typically myeloablative or reduced-intensity, built around busulfan, fludarabine, or treosulfan. The decision of when to transplant is individualised: the child with good infection control on prophylaxis may defer, while the child with recurrent severe infection or intractable inflammatory complications benefits from earlier transplant. [7]

Gene therapy is an emerging curative option for CGD, using a lentiviral or gamma-retroviral vector to insert a corrected copy of the defective gene (gp91phox for X-linked, p47phox for the commonest autosomal recessive form) into the patient's own haematopoietic stem cells. Because the cells are the patient's own, there is no graft-versus-host disease and no need for a matched donor, which is attractive for the CGD patient without a matched sibling. Early gamma-retroviral trials were limited by transient correction and, in some cases, insertional oncogenesis, but the newer lentiviral vectors with improved safety profiles are under active investigation and have shown promising short-term oxidase reconstitution in expert centres. Gene therapy is not yet the standard of care, but it is the direction of travel for CGD and the candidate should know that it exists and why. [4] [5]
For severe congenital neutropenia, the long-term management is G-CSF to maintain the neutrophil count and serial bone-marrow surveillance — aspirate, biopsy, and cytogenetics — at least annually to detect the clonal evolution that heralds myelodysplasia or leukaemia. The cumulative risk of myelodysplasia or acute myeloid leukaemia is roughly five percent per year in some cohorts, and the detection of a monosomy 7 or an acquired trisomy 21 clone is the trigger for urgent transplant evaluation. HSCT is the only curative therapy for congenital neutropenia and is offered when clonal evolution appears or when the infection burden is unmanageable despite G-CSF. [10]
Long-term follow-up after transplant for any phagocyte disorder monitors immune reconstitution — the recovery of phagocyte function, confirmed by a normal DHR assay after CGD transplant — manages graft-versus-host disease, and maintains infection and malignancy surveillance. For the Chédiak-Higashi child, the transplant cures the haematological and immune phenotype but does not fully arrest the progressive neurological involvement, so neurological follow-up continues. The family receives genetic counselling, carrier testing for X-linked CGD, and the offer of prenatal or preimplantation genetic diagnosis for future pregnancies. The arc of CGD — from a disease of recurrent life-threatening infection to one in which transplant offers cure — is one of the successes of modern paediatric immunology, and it rests on the twin pillars of antimicrobial prophylaxis and curative transplantation. [7] [12]
Specific Subtypes & Scenarios
X-linked CGD (CYBB / gp91phox) is the commonest form and the prototype. A boy with recurrent abscess-forming infection by the CGD cluster, granulomatous complications, and a virtually absent DHR fluorescence has X-linked CGD until the CYBB mutation is confirmed. Maternal carrier testing is available, and carrier detection by flow cytometry shows the mosaic (two-population) pattern of X-chromosome inactivation in carrier females. Prenatal and preimplantation genetic diagnosis allows family planning. Gene therapy and HSCT are the curative options, with prophylaxis as the bridge. [4] [6]
Autosomal recessive p47phox CGD (NCF1) is the second commonest form and accounts for about a quarter of all CGD. It is typically milder than the X-linked form, because the residual oxidase activity in some cells produces a partial oxidative burst, and the DHR assay shows a reduced-but-present fluorescence rather than the flat line of X-linked disease. The Kuhns study established that p47phox deficiency arises almost exclusively from the recombination event between NCF1 and its pseudogenes, a recurrent mutational mechanism. Girls and boys are equally affected. The management — prophylaxis and transplant — is the same. [6]
Leukocyte adhesion deficiency type 1 (ITGB2 / CD18) is the adhesion defect the examination tests. The neonatal triad of delayed cord separation, omphalitis without pus, and a striking leukocytosis is the presentation, and flow cytometry for CD18 confirms it. The severe form presents in infancy with life-threatening infection; the moderate form presents later with periodontal disease and recurrent skin infection. HSCT is curative. The candidate must be able to explain why the neutrophil count is high — because the cells are made normally but cannot leave the bloodstream — and why there is no pus — because pus is neutrophils in tissue, and in LAD the neutrophils are stuck in the vessels. [8] [9]
Severe congenital neutropenia (ELANE, HAX1, G6PC3) is the production defect that presents with overwhelming bacterial sepsis in early infancy. ELANE (autosomal dominant) is the commonest and accounts for about half of cases; HAX1 (autosomal recessive) adds a neurological phenotype; G6PC3 and the rarer genes complete the genetic spectrum. G-CSF is the mainstay, and serial bone-marrow cytogenetics is mandatory for the myelodysplasia and leukaemia surveillance. Cyclic neutropenia, also from ELANE, runs the 21-day cycle and is generally milder. HSCT is curative and is offered for clonal evolution or refractory infection. [10]
Chédiak-Higashi syndrome (LYST) is the granule-packaging defect with the unforgettable blood film. Giant intracytoplasmic granules in all granule-bearing cells, partial oculocutaneous albinism, a mild bleeding tendency, recurrent pyogenic infection, and progressive neurological involvement make up the phenotype, and the accelerated (haemophagocytic) phase is the chief cause of death. The accelerated phase presents with fever, hepatosplenomegaly, lymphadenopathy, and pancytopenia, and demands urgent haematology-led treatment with the HLH chemotherapy protocol as a bridge to curative transplant. HSCT cures the haematological and immune phenotype but does not arrest the neurological progression. [11]
Specific granule deficiency (C/EBPe) and Shwachman-Diamond syndrome (SBDS) are rarer members of the family. Specific granule deficiency, from C/EBPe, produces atypical infection from the absence of the neutrophil-specific granule contents and is distinguished by the absence of specific granules on electron microscopy. Shwachman-Diamond syndrome combines neutropenia (the dominant haematological feature), pancreatic exocrine insufficiency causing steatorrhoea and failure to thrive, skeletal changes (metaphyseal chondrodysplasia), and a risk of myelodysplasia and leukaemia. Myeloperoxidase deficiency (MPO), the commonest neutrophil defect by frequency, is usually asymptomatic and is mentioned only because it produces a persistently negative NBT or DHR variant in an otherwise well person — clinically relevant mainly in diabetic patients who may develop candidiasis. [3] [10]
Complications & Pitfalls
The chief avoidable cause of death across the phagocyte disorders is delayed recognition of the infecting organism cluster. A child with a staphylococcal liver abscess or an invasive Aspergillus pneumonia who is treated episode-by-episode, without the DHR assay being ordered, is the child who accumulates organ damage and presents for transplant later and sicker. The candidate who asks which organism caused each infection, and who orders the DHR assay when the cluster fits, is the one who makes the diagnosis in time. The organism list is the single most sensitive screening tool for CGD. [4] [5]
The live-vaccine pitfall is preventable. The BCG vaccine, safe in an immunocompetent child, can cause local or disseminated disease in CGD and LAD because the attenuated mycobacterium persists in a defective phagocyte. In countries that give neonatal BCG, a non-healing BCG-site ulcer or regional lymphadenitis in a child who later proves to have CGD is a sentinel complication, and the rule is to withhold BCG while a severe phagocyte disorder is being investigated. The Shearer and IDF recommendations set out the live-vaccine approach for immunodeficient patients and their close contacts. [12]
The inflammation pitfall is the failure to recognise that CGD is a disease of both infection and inflammation. The granulomatous colitis, the gastric-outlet obstruction, and the genitourinary obstruction are not ordinary inflammatory bowel disease or ordinary surgical obstruction — they are dysregulated granulomatous inflammation driven by the killing defect, and they require anti-inflammatory therapy (often corticosteroids) alongside the antimicrobials and the prophylaxis. The candidate who treats only the infection misses half the disease. [4]
The myelodysplasia pitfall in severe congenital neutropenia is the failure to maintain serial bone-marrow surveillance. The cumulative risk of myelodysplasia or acute myeloid leukaemia is substantial, and the detection of a monosomy 7 or an acquired trisomy 21 clone on serial cytogenetics is the trigger for urgent transplant evaluation. A child with congenital neutropenia who develops new cytopenias, macrocytosis, or a cytogenetic clone is evolving to myelodysplasia or leukaemia, and the surveillance cannot lapse. [10]
The accelerated-phase pitfall in Chédiak-Higashi is the failure to recognise that a fever with hepatosplenomegaly in a child with the syndrome is the start of the often-fatal haemophagocytic phase. The accelerated phase demands emergency haematology involvement and the HLH protocol as a bridge to transplant, and delay is lethal. The transplant complications — graft-versus-host disease, failure of engraftment, and the post-transplant infections that complicate any conditioning regimen — determine the long-term outcome and are the focus of post-transplant care across all the phagocyte disorders. [7] [11]
Prognosis & Disposition
Untreated CGD, before the era of prophylaxis and transplant, carried a mortality of roughly two to five percent per year and a reduced lifespan driven by cumulative organ damage from recurrent infection and inflammation. Modern management — co-trimoxazole and anti-mould prophylaxis, aggressive treatment of acute infection, and the option of curative transplant — has transformed the prognosis. The Chiesa 712-patient cohort established overall survival after HSCT for CGD at roughly 70 to 95 percent across genotypes and ages, with the best outcomes in children transplanted before severe infection-driven organ damage and with a matched sibling donor. Prophylaxis alone, in well-controlled children, supports survival into adulthood, and the decision to transplant is individualised. [4] [7]
For LAD-1, the severe form has a poor prognosis without transplant, with significant mortality in early childhood from intractable infection; HSCT is curative and is offered early in the severe phenotype. For severe congenital neutropenia, G-CSF transforms the infection risk and supports near-normal survival in childhood, but the long-term myelodysplasia and leukaemia risk persists and determines the eventual need for transplant. For Chédiak-Higashi syndrome, the prognosis is dominated by the accelerated phase — the chief cause of death — and by the progressive neurological involvement that transplant does not arrest; HSCT cures the haematological phenotype and prevents the accelerated phase, but the neurological course continues. [8] [10] [11]
Every child with a confirmed severe phagocyte disorder is referred to a paediatric immunology and stem cell transplant centre for ongoing care and transplant evaluation. The family receives genetic counselling, carrier testing for X-linked CGD, and the offer of prenatal or preimplantation genetic diagnosis for future pregnancies. The long-term survivor — whether transplanted or managed medically — enters a transition programme to adult immunology and infectious diseases care, with attention to infection surveillance, inflammation control, malignancy surveillance, reproductive and genetic counselling, and the psychosocial adjustment to a chronic immunodeficiency. The arc of the disease, from recurrent life-threatening infection to cure or durable control, rests on the twin pillars of prophylaxis and curative transplantation. [4] [12]
Special Populations
In countries with universal newborn screening for severe combined immunodeficiency (TREC), the phagocyte disorders are not detected by the screen — the T-cell count is normal — so the clinician must suspect the diagnosis from the clinical picture. The CGD organism cluster, the LAD neonatal triad, the profound neutropenia, and the Chédiak-Higashi blood film are the front-line tools, and the DHR and flow cytometry assays are the confirmatory tests. In countries without ready access to flow cytometry, the diagnosis is delayed and the infectious burden accumulates, which is why equitable access to immunological testing is an advocacy priority. [4] [6]
Families with a known CGD mutation, particularly the X-linked form, are offered maternal carrier testing, sibling testing, and prenatal or preimplantation genetic diagnosis for future pregnancies. The power of early diagnosis is that prophylaxis can begin before the first severe infection and transplant can be planned before organ damage accumulates, so the reproductive counselling is part of the management. The family that has lost a child to CGD, and that is offered prenatal diagnosis for the next pregnancy, is the family that may see their next child managed from birth and transplanted before severe infection. [6] [12]
For Aboriginal and Torres Strait Islander, Maori, and other Indigenous children, and for refugee, asylum-seeking, and migrant families, the barriers to immunological testing, prophylaxis, and transplant services must be addressed explicitly. Consanguinity increases the rate of the autosomal recessive phagocyte disorders, which is relevant in some migrant and refugee populations. Culturally safe genetic counselling, with trained interpreters, and equitable access to the transplant pathway are essential. The BCG-vaccine issue is especially relevant in countries that immunise at birth and in migrant families who received BCG overseas, because a non-healing BCG site may be the first clue to CGD or LAD. [5] [12]
In rural and remote settings, early recognition of the CGD organism cluster and the LAD neonatal triad, early retrieval to a transplant centre, and telehealth immunology and infectious diseases support are the front line. The family must understand that a phagocyte disorder is a serious condition, that prophylaxis cannot lapse, and that the protections — no live BCG, irradiated blood if transfusion is needed, urgent referral for severe infection — cannot wait for the next routine appointment. The rural clinician who suspects CGD, orders the DHR assay, starts co-trimoxazole and anti-mould prophylaxis, and arranges the immunology referral is the clinician who changes the outcome. [4]
Adolescents with phagocyte disorders transitioning to adult care — transplanted CGD and LAD patients entering adulthood, congenital neutropenia survivors on long-term G-CSF, and Chédiak-Higashi survivors with ongoing neurological disease — need a structured transition that addresses infection and malignancy surveillance, inflammation control, reproductive and genetic counselling, and the psychosocial adjustment to a chronic immunodeficiency. The transition is not a handover but a planned programme, and its quality determines whether the gains of prophylaxis and early transplant are sustained into adult life. [7] [10]
Evidence, Guidelines & Regional Differences
The IUIS classification, updated in 2024 by the Poli committee and previously in the Bousfiha phenotypic classification of 2017 and the Tangye interim update of 2021, is the authoritative taxonomic reference for the phagocyte disorders, grouping them under congenital defects of phagocyte number or function. Together these references define the modern landscape of the field and the place of CGD, LAD, the neutropenias, and the granule disorders within it. [1] [2] [3]
The CGD evidence is anchored by three references the candidate must know. The Holland review (2013) is the comprehensive clinical reference, setting out the organism cluster, the pathophysiology of the respiratory burst, and the granulomatous complications. The Arnold and Heimall review (2017) is the modern management reference, establishing co-trimoxazole and anti-mould prophylaxis as the standard of care. The Chiesa 712-patient multicentre cohort (Blood 2020) is the transplant-outcomes evidence, confirming 70 to 95 percent overall survival after HSCT across genotypes and ages and establishing the principle that transplant before organ damage achieves the best results. The Kuhns study (Blood Advances 2019) is the genetic and diagnostic reference, establishing the p47phox pseudogene recombination mechanism and the modern molecular diagnostics. [4] [5] [6] [7]
For LAD, the van de Vijver review (Hematology/Oncology Clinics 2013) and the Hanna and Etzioni review (Annals of the New York Academy of Sciences 2012) together set out the three-type framework and the molecular basis of each. For the congenital neutropenias, the Skokowa primer (Nature Reviews Disease Primers 2017) is the comprehensive reference, covering the genetics, the pathophysiology of the unfolded-protein response, the G-CSF management, and the myelodysplasia surveillance. For Chédiak-Higashi, the Talbert, Malicdan, and Introne review (Current Opinion in Hematology 2023) sets out the modern understanding of the granule-trafficking defect and the accelerated phase. [8] [9] [10] [11]
The live-vaccine caution is set out in the recommendations of the Medical Advisory Committee of the Immune Deficiency Foundation, published by Shearer and colleagues in 2014, which remain the standard reference for vaccination in immunodeficient patients. Regional differences in the vaccine schedule — in particular, whether neonatal BCG is given — determine the vaccine complications a clinician in a given country is likely to see, and the non-healing BCG site is a sentinel clue in BCG-vaccinating countries. [12]
The active controversies include the optimal timing of transplant in CGD (immediately on diagnosis versus after a period of prophylaxis-driven stability), the role and long-term safety of gene therapy (including the insertional oncogenesis events in the early gamma-retroviral trials that led to the development of safer lentiviral vectors), the role of interferon-gamma in regions where mould-active antifungal prophylaxis is standard, and equitable access to prophylaxis and transplant globally. The candidate should be able to discuss these controversies at viva level, defending the evidence for each position. [4] [7]
Exam Pearls
Chronic granulomatous disease is the prototype phagocyte disorder — a defective NADPH oxidase abolishes the respiratory burst, so catalase-positive organisms survive and the host responds with granulomatous inflammation. The bedside clue is the organism cluster: Staphylococcus aureus, Burkholderia, Serratia, Nocardia, and Aspergillus. [4] [5]
The respiratory burst is elegant: the phagocyte generates hydrogen peroxide via NADPH oxidase, and myeloperoxidase uses that peroxide plus a halide to make bleach. Catalase-positive organisms destroy their own peroxide, so they survive only when the phagocyte cannot make its own — this is why CGD patients get staphylococcal and Aspergillus infection but not streptococcal. [4]
The dihydrorhodamine (DHR) flow cytometry assay is the diagnostic test for CGD — it quantifies the oxidative burst, distinguishes the flat line of X-linked disease from the reduced-but-present pattern of p47phox, and has replaced the nitroblue tetrazolium test. The neutrophil count is normal in CGD, so a normal count never excludes the disease. [4] [6]
CGD genotypes: X-linked CYBB (gp91phox) is two-thirds of cases; autosomal recessive p47phox (NCF1) is a quarter and arises from recombination with its pseudogenes; p67phox, p22phox, and p40phox complete the spectrum. [6]
Leukocyte adhesion deficiency type 1 (ITGB2 / CD18) is delayed cord separation, omphalitis without pus, and a striking leukocytosis — the cells are made normally and the burst is intact, but without CD18 the neutrophils cannot emigrate from the vessel. Flow cytometry for CD18 confirms it, and HSCT is curative. [8] [9]
Severe congenital neutropenia (ELANE commonest, also HAX1, G6PC3) presents with overwhelming bacterial sepsis in early infancy, is managed with G-CSF and serial marrow cytogenetics, and carries a cumulative myelodysplasia and leukaemia risk that mandates surveillance. Cyclic neutropenia runs the 21-day cycle. [10]
Chédiak-Higashi syndrome (LYST) is giant granules, partial albinism, a bleeding tendency, recurrent pyogenic infection, and a fatal accelerated (haemophagocytic) phase — HSCT cures the haematological phenotype but not the progressive neurology. [11]
Co-trimoxazole plus an anti-mould antifungal (itraconazole or posaconazole) is the prophylaxis backbone of CGD, and the BCG vaccine is withheld from children with severe phagocyte defects. HSCT achieves 70 to 95 percent overall survival in CGD (Chiesa 712-patient cohort), and gene therapy is the emerging curative option. [5] [7] [12]
References
- [1]Poli MC; Aksentijevich I; Bousfiha AA; Cunningham-Rundles C; Hambleton S; Klein C; Morio T; Picard C Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Hum Immun, 2025.PMID 41608114
- [2]Tangye SG; Al-Herz W; Bousfiha A; Cunningham-Rundles C; Franco JL; Holland SM; Klein C; Morio T The Ever-Increasing Array of Novel Inborn Errors of Immunity: an Interim Update by the IUIS Committee. J Clin Immunol, 2021.PMID 33598806
- [3]Bousfiha A; Jeddane L; Picard C; Ailal F; Bobby Gaspar H; Al-Herz W; Chatila T; Crow YJ The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol, 2018.PMID 29226301
- [4]Holland SM Chronic granulomatous disease. Hematol Oncol Clin North Am, 2013.PMID 23351990
- [5]Arnold DE; Heimall JR A Review of Chronic Granulomatous Disease. Adv Ther, 2017.PMID 29168144
- [6]Kuhns DB; Hsu AP; Sun D; Lau K; Fink D; Griffith P; Shapiro RA; Malech HL NCF1 (p47(phox))-deficient chronic granulomatous disease: comprehensive genetic and flow cytometric analysis. Blood Adv, 2019.PMID 30651282
- [7]Chiesa R; Wang J; Blok HJ; Hazelaar S; Neven B; Moshous D; Friedacher K; Köglmeier J; Qasim W Hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults. Blood, 2020.PMID 32614953
- [8]van de Vijver E; van den Berg TK; Kuijpers TW Leukocyte adhesion deficiencies. Hematol Oncol Clin North Am, 2013.PMID 23351991
- [9]Hanna S; Etzioni A Leukocyte adhesion deficiencies. Ann N Y Acad Sci, 2012.PMID 22276660
- [10]Skokowa J; Dale DC; Touw IP; Zeidler C; Welte K Severe congenital neutropenias. Nat Rev Dis Primers, 2017.PMID 28593997
- [11]Talbert ML; Malicdan MCV; Introne WJ Chediak-Higashi syndrome. Curr Opin Hematol, 2023.PMID 37254856
- [12]Medical Advisory Committee of the Immune Deficiency Foundation; Shearer WT; Fleisher TA; Buckley RH; Ballas Z; Ballow M Recommendations for live viral and bacterial vaccines in immunodeficient patients and their close contacts. J Allergy Clin Immunol, 2014.PMID 24582311