Paeds · allergy-and-immunology
Primary immunodeficiency: warning signs and diagnostic approach
Also known as Inborn errors of immunity · Primary immunodeficiency disease · Recurrent infection workup · Jeffrey Modell warning signs · PID screening
Fellowship topic on primary immunodeficiency (inborn errors of immunity) in children: the Jeffrey Modell ten warning signs as the screening tool that turns 'just recurrent infections' into a referral; the modern IUIS 2022 classification spanning cellular-humoral, syndromic, antibody, immune-dysregulation, phagocyte, innate, autoinflammatory, complement, bone-marrow-failure and phenocopy categories; the pathophysiological logic that maps each immune arm to a characteristic infection signature — T-cell deficiency to opportunistic and viral or fungal infection, antibody deficiency to recurrent sinopulmonary infection with encapsulated bacteria, phagocyte defects to catalase-positive abscesses, and complement deficiency to neisserial sepsis; the tiered diagnostic protocol of screening immunoglobulins and full blood count with differential, escalating to lymphocyte-subset flow cytometry, vaccine-antibody response, CH50/AP50, neutrophil-function testing, TREC/KREC and genetic sequencing; the time-critical SCID prototype where lymphopenia and failure to thrive demand same-day immunology referral and a move to haematopoietic stem cell transplant before infection; the regional realities of newborn SCID screening by TREC; and the management stance of immunoglobulin replacement, infection prophylaxis, vaccination strategy, genetic counselling and the avoidance of live vaccines until cleared.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture an eight-month-old boy brought to clinic because he is not growing, has had two pneumonias in three months needing intravenous antibiotics, and has persistent oral thrush that will not clear. His full blood count shows a lymphocyte count that is low for his age. A cousin on the father's side died in infancy of an infection the family never understood. That cluster — failure to thrive, persistent opportunistic mucosal infection, recurrent deep infection needing intravenous antibiotics, lymphopenia, and a family history of early childhood death — is the signature that should make a clinician stop treating infection after infection and ask whether the child has a primary immunodeficiency. [1] [5]
Primary immunodeficiency is the older name for what the field now calls inborn errors of immunity: inherited disorders in which a defect in the development, function or regulation of the immune system produces a characteristic susceptibility to infection, immune dysregulation, autoimmunity, malignancy or a syndromic phenotype. They are not one disease but a large and growing family — the 2022 International Union of Immunological Societies classification lists nearly five hundred distinct conditions across ten categories — and the clinical challenge is that most children who present with recurrent infection do not have one. The skill the fellowship examiner is testing is the judgement to distinguish ordinary, exposure-driven childhood infection from the pattern that crosses into an immune-deficiency signature, and to act on that judgement with a tiered workup rather than another antibiotic course. [2] [3]
Classification
Primary immunodeficiencies are classified in two complementary ways: by the laboratory immune-arm affected, and by the clinical pattern of infection. Both matter, because the first directs the diagnostic workup and the second directs the bedside suspicion. The International Union of Immunological Societies 2022 phenotypic classification groups inborn errors of immunity into ten categories, and knowing the shape of these categories is how a candidate anchors an answer rather than reciting a list. The first category is immunodeficiencies affecting cellular and humoral immunity — the combined immunodeficiencies, of which severe combined immunodeficiency is the prototype. The second is combined immunodefencies with syndromic features, such as Wiskott–Aldrich syndrome, ataxia-telangiectasia and CHARGE syndrome, where the immune defect travels with a recognisable physical or developmental phenotype. [3] [4]

The third category, predominantly antibody deficiencies, is the largest in practice and includes X-linked agammaglobulinaemia, common variable immunodeficiency and selective immunoglobulin-A deficiency — the group that presents with recurrent sinopulmonary infection. The fourth is diseases of immune dysregulation, including autoimmune lymphoproliferative syndrome, IPEX, APECED and the haemophagocytic syndromes, where the immune system fails to regulate itself and the child presents with autoimmunity or uncontrolled inflammation rather than infection alone. The fifth category, congenital defects of phagocyte number or function, includes chronic granulomatous disease, leucocyte adhesion deficiency and severe congenital neutropenia. The remaining categories cover defects in innate immunity, autoinflammatory disorders, complement deficiencies, bone-marrow failure and cytopenias, and the phenocopies of primary immunodeficiency such as somatic or autoantibody-driven variants. The practical point for the candidate is that the category tells you which infection signature to expect and which laboratory arm to interrogate. [2] [4]
Epidemiology & Risk Factors
Where does primary immunodeficiency sit in the workload of a general paediatrician, and who is the child at risk? The overall prevalence of diagnosed inborn errors of immunity has risen over the past two decades, partly through better recognition and newborn screening and partly through the expanding classification. United States data spanning 2001 to 2007 estimated a diagnosed prevalence in the range of one in two thousand children, with antibody deficiencies the most commonly identified group and a meaningful share of children hospitalised or disabled by their condition. The point the examiner wants is that these disorders are uncommon enough to be missed and common enough that every general paediatrician will see at least one — the danger is the false reassurance that 'recurrent infection is normal in children'. [10]
The risk factors that should raise the index of suspicion are a family history of primary immunodeficiency or of early childhood death from infection, consanguinity (which increases the chance of autosomal-recessive forms), male sex for the X-linked conditions such as X-linked agammaglobulinaemia and X-linked SCID, and the characteristic infection patterns described below. The fatal and severe case profile clusters in infants with combined immunodeficiency who are diagnosed late — after they have already acquired opportunistic infection — which is exactly the harm that newborn TREC screening is designed to prevent. The longitudinal data linking newborn screening to survival after haematopoietic cell transplantation for SCID show that earlier detection, before infection, is the single most powerful determinant of outcome. [6] [11]
Pathophysiology
Why does a defect in a single immune component produce a signature pattern of infection, and why does that signature matter at the bedside? The answer is that the immune system is organised into arms — T-cell and combined, antibody, phagocyte, complement and innate — and each arm has a restricted set of pathogens it is built to clear. When one arm fails, the child does not become generally 'prone to infection'; the child becomes prone to a specific, recognisable subset of organisms, and that subset is the diagnostic clue. The pathophysiological logic, set out in the antibody-deficiency and broader reviews, is that the pattern of organism points to the arm, and the arm points to the test. [7]
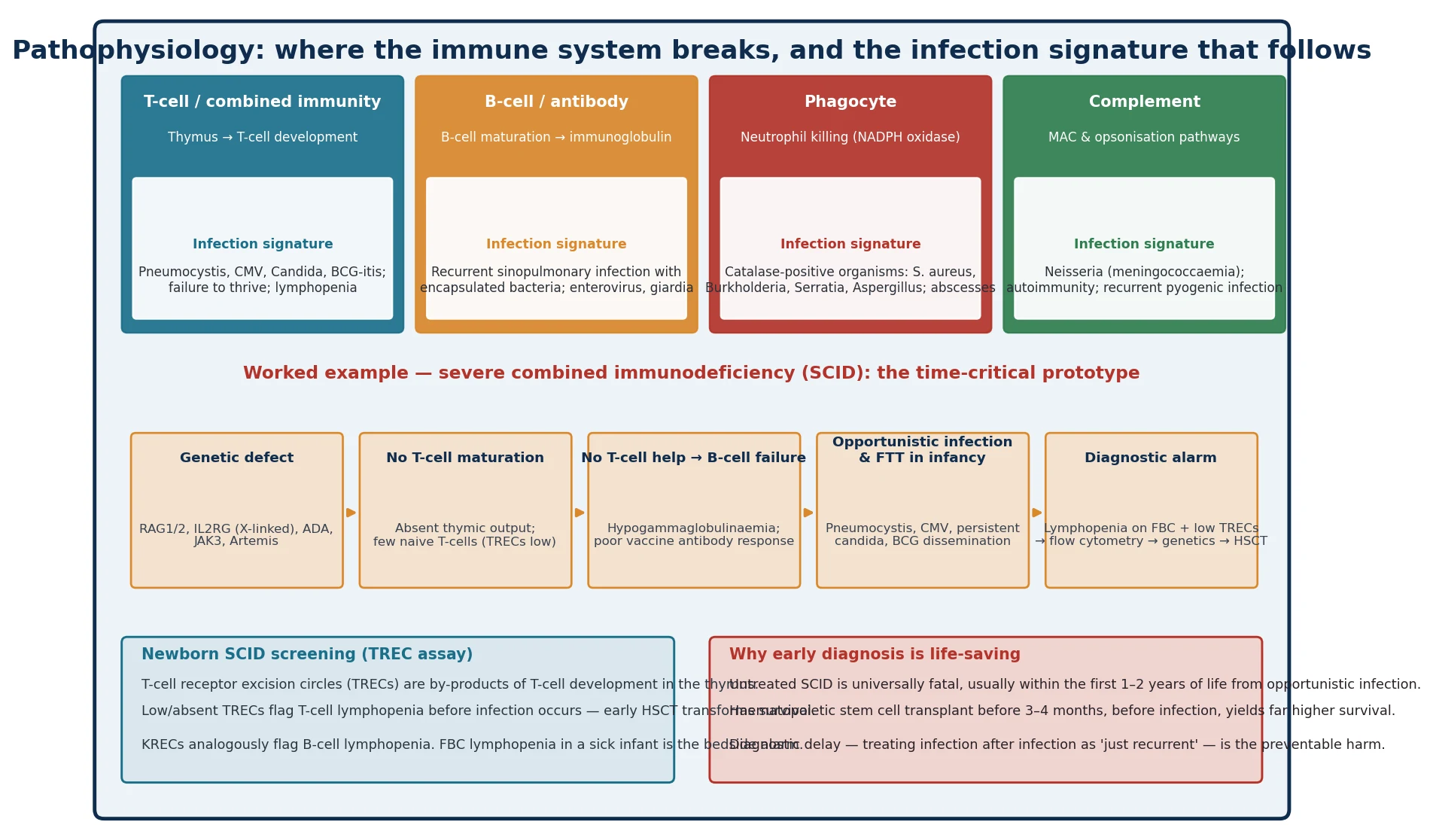
The T-cell and combined arm governs cell-mediated immunity. When it fails, the child cannot clear intracellular, viral and fungal pathogens, so the signature is opportunistic infection — Pneumocystis jirovecii pneumonia, cytomegalovirus, persistent Candida, disseminated bacille Calmette–Guérin after vaccination — together with failure to thrive and lymphopenia. Severe combined immunodeficiency is the extreme: a genetic defect in T-cell development (IL2RG in the X-linked form, RAG1/2, adenosine deaminase, JAK3, Artemis) leaves the thymus unable to produce naive T-cells, and because T-cells provide help to B-cells, antibody production fails too — hence 'combined'. The diagnosis is a clinical emergency, because untreated SCID is universally fatal within the first one to two years of life. [9] [12]
The antibody arm governs humoral immunity against encapsulated extracellular bacteria. When it fails — in X-linked agammaglobulinaemia, common variable immunodeficiency or a significant antibody deficiency — the signature is recurrent sinopulmonary infection with Streptococcus pneumoniae and Haemophilus influenzae, bronchiectasis from repeated damage, and susceptibility to enteroviral and giardial infection. The phagocyte arm governs intracellular killing through the NADPH-oxidase respiratory burst. When it fails, in chronic granulomatous disease, the signature is recurrent deep abscess and granulomatous inflammation with catalase-positive organisms — Staphylococcus aureus, Burkholderia cepacia, Serratia, Nocardia and Aspergillus. The complement arm governs opsonisation and the membrane-attack complex; when it fails, especially in the terminal pathway, the signature is invasive Neisseria disease — meningococcaemia — and a tendency to autoimmunity. [7]
Clinical Presentation
The presentation of primary immunodeficiency is, with the exception of the acutely unwell SCID infant, chronic and recurrent rather than a single acute event, and the clinical skill is synthesising the pattern over time. The Jeffrey Modell Foundation ten warning signs are the bedside tool: four or more new ear infections within a year, two or more serious sinus or lung infections within a year, two or more months on antibiotics with little effect, two or more deep-seated skin or organ infections, failure to thrive in an infant, recurrent deep abscesses, persistent oral thrush or cutaneous candidiasis beyond infancy, the need for intravenous antibiotics to clear infections, two or more deep-seated infections, and a family history of primary immunodeficiency. The United Kingdom prospective study that tested these signs in a paediatric immunology population found that the number of warning signs correlated with the likelihood of a confirmed primary immunodeficiency, though no single sign is perfectly sensitive or specific — which is why the signs are a prompt to investigate, not a diagnostic rule. [1]
The syndromic presentations are equally important and often the clue that converts 'recurrent infection' into a named condition. An infant boy with recurrent infection, eczema, thrombocytopenia and small platelets has Wiskott–Aldrich syndrome. A child with recurrent sinopulmonary infection and progressive cerebellar ataxia, oculocutaneous telangiectasia and elevated alpha-fetoprotein has ataxia-telangiectasia. A boy with recurrent, severe bacterial infection from infancy and absent tonsillar tissue has X-linked agammaglobulinaemia. A child with recurrent lymphadenitis, deep abscesses, osteomyelitis and granulomatous skin lesions with a positive nitroblue-tetrazolium or dihydrorhodamine test has chronic granulomatous disease. The developmental or morphological feature is often the first thing the family notices, and the recurrent infection is what brings the child to medical attention — synthesising the two is the examiner's expectation. [2] [7]
MR. POOR — warning signs that earn a workup
Differential Diagnosis
The differential diagnosis of recurrent infection in a child is broad, and the central pitfall is the mirror image of the missed immunodeficiency: over-investigating the normal child, or failing to exclude secondary and non-immune causes before labelling the pattern 'primary'. The commonest explanation for frequent infection in a young child is normal immune maturation under high exposure — daycare attendance, siblings, winter viral seasons — producing six to ten upper-respiratory infections a year with normal growth, normal examination between episodes and no serious infections. Anatomical causes include obstructive sleep-disordered breathing with recurrent otitis, or structural lung disease. Atopic disease — allergic rhinitis and eczema — predisposes to recurrent upper-respiratory and skin infection and is far commoner than primary immunodeficiency. [5]
Secondary immunodeficiency must be excluded before a primary cause is assigned, because treating the cause reverses the susceptibility. Human immunodeficiency virus, protein loss through nephrotic syndrome or protein-losing enteropathy, immunosuppressive therapy including steroids and biologics, malignancy and chemotherapy, and malnutrition all impair immunity and produce recurrent infection. The diagnostic protocol from the European Society for Immunodeficiencies builds an explicit HIV test into the second-line workup, and the candidate should always ask about medication, nutrition, and gastrointestinal and renal loss before settling on an inherited cause. The practical rule is that a child with normal growth, normal examination, no deep or unusual infections and no family history is overwhelmingly likely to have normal exposure-driven infection, whereas the child who breaks any one of those rules deserves the tiered workup. [5] [8]
Clinical & Bedside Assessment
The bedside assessment of a child referred for recurrent infection is structured around three questions: is the infection pattern normal, is there a non-immune cause, and is there a signature that crosses into immunodeficiency? The history establishes the frequency, severity, persistence and organism of infections — how many, how deep, how long, what was isolated, what antibiotics were needed and whether they worked — alongside growth, developmental and syndromic features, family history including consanguinity and early childhood death, and the child's medication and nutritional status. The frequency thresholds of the warning signs are the operational test: four or more ear infections, two or more serious sinus or lung infections, two or more months on antibiotics with little effect. [1]
The examination looks for the syndromic and immunological clues. Growth parameters and their trajectory identify failure to thrive, the cardinal sign of combined immunodeficiency. The mouth and skin are inspected for persistent candidiasis; the lymphoid tissue for absent tonsils, suggesting X-linked agammaglobulinaemia, or generalised lymphadenopathy and hepatosplenomegaly suggesting immune dysregulation. The skin is examined for eczema and petechiae of Wiskott–Aldrich, the telangiectasia of ataxia-telangiectasia, and the granulomatous and abscess scars of chronic granulomatous disease. The chest is auscultated for the crackles and wheeze of bronchiectasis. The vital signs identify the acutely unwell infant who needs escalation now, and the development and dysmorphism are screened for the syndromic associations. The assessment ends with a clear statement of which warning signs are present and which first-line tests will be ordered. [8]
Investigations
The investigation of suspected primary immunodeficiency is deliberately tiered, so that a non-immunologist can perform the first stage and so that expensive and specialist tests are reserved for the children who need them. The European Society for Immunodeficiencies multi-stage protocol defines the structure, and the candidate should present the investigations as a ladder, not a list. Tier one, for the primary-care or general-paediatric setting, is the full blood count with differential — paying particular attention to the absolute lymphocyte and neutrophil counts, age-appropriate because lymphocyte counts are high in infancy — and serum immunoglobulins IgG, IgA and IgM. A vaccine-antibody response, classically to tetanus, pneumococcus or Haemophilus, is added if the history suggests antibody deficiency. An HIV test is offered to exclude secondary immunodeficiency. [5]
Tier two, in the general-paediatric or immunology setting, adds lymphocyte-subset flow cytometry — CD3, CD4, CD8, CD19 and CD56 — to quantify T, B and natural-killer cells, and a formal vaccine-antibody response measured before and after a vaccine challenge to test functional antibody production. Where SCID or another combined deficiency is suspected, naive and memory T-cell markers (CD45RA and CD45RO) and TREC analysis are added. Tier three, in the specialist immunology service, adds neutrophil-function testing — the dihydrorhodamine or nitroblue-tetrazolium assay for chronic granulomatous disease — complement assays CH50 and AP50 followed by individual components, KREC analysis for B-cell lymphopenia, and genetic testing by targeted gene panel or whole-exome sequencing to confirm a molecular diagnosis and enable family counselling. Newborn screening for SCID by the TREC assay, now established across many high-income programmes, is the population-level tier-zero test that identifies T-cell lymphopenia before infection occurs. [5] [6]
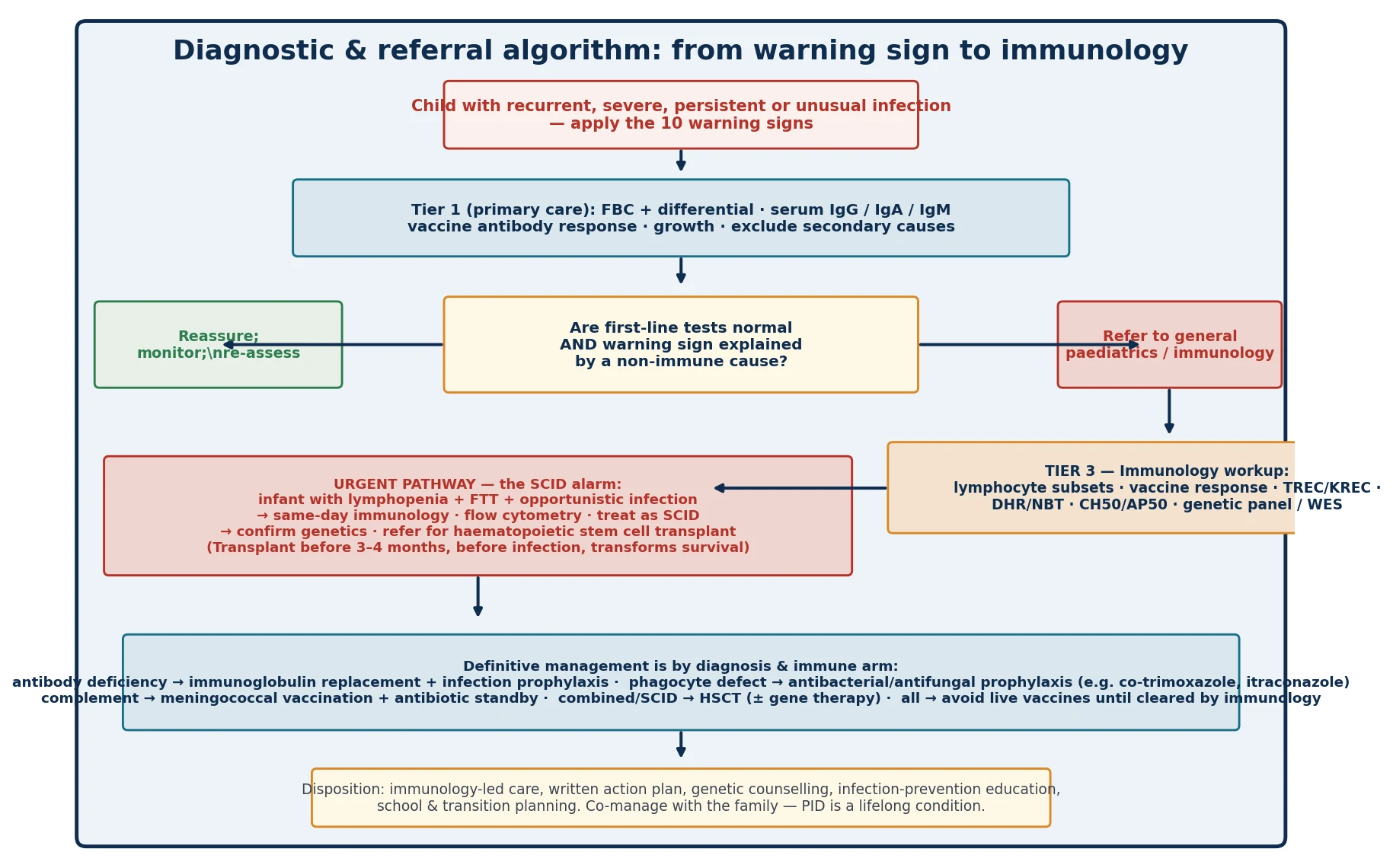
The interpretation requires age-specific reference ranges, because the normal infant has a far higher absolute lymphocyte count than an older child or adult, so a lymphocyte count that looks 'normal' at an adult threshold may in fact be lymphopenic for age — the bedside reason SCID is missed. The candidate should be able to state that a normal immunoglobulin level does not exclude a functional antibody deficiency, which is why the vaccine-antibody response is the functional test, and that a normal total lymphocyte count does not exclude a T-cell defect if the subsets are abnormal. The ESID working definitions synthesise these results into operational clinical diagnostic criteria for the major inborn errors of immunity, and they are the reference point for a defensible diagnostic conclusion. [8] [12]
Management — Resuscitation
The resuscitation requirement in primary immunodeficiency applies almost entirely to two scenarios: the acutely septic child, and the infant with suspected or confirmed SCID. For the acutely septic child, the management is standard paediatric sepsis care — oxygen, fluid resuscitation and broad-spectrum intravenous antibiotics — but with two specific additions. First, the organism and its pattern guide empiric cover, so a child with known or suspected complement deficiency who presents with fever is managed as presumptive meningococcaemia and receives a third-generation cephalosporin. Second, once a primary immunodeficiency is known or suspected, the antibiotic choice is coordinated with immunology, because some defects require specific prophylaxis and some live investigations, such as certain vaccines, are contraindicated. [8]
For the infant with suspected SCID — the lymphopenic, failing-to-thrive baby with opportunistic infection — the resuscitation is protective as much as it is curative. The child is kept in isolation, prophylactic antibiotics are started against Pneumocystis (typically co-trimoxazole), antiviral and antifungal prophylaxis is added as guided by the immunology team, live vaccines are withheld, blood products are given only irradiated and cytomegalovirus-safe, and breastfeeding is continued only if the mother is cytomegalovirus-negative. The same-day immunology referral and the move to haematopoietic stem cell transplantation before infection are the definitive acts, and everything in the resuscitation phase is designed to keep the child infection-free until transplant can occur. [9] [11]
Management — Definitive & Stepwise
The definitive management of primary immunodeficiency is by diagnosis and immune arm, and the candidate should structure the answer by category rather than reciting individual drugs. For the predominantly antibody deficiencies, the cornerstone is immunoglobulin replacement therapy — intravenous or subcutaneous — with the aim of keeping trough immunoglobulin-G levels protective and preventing the bronchiectasis and end-organ damage that follow recurrent infection. Antibiotic prophylaxis and aggressive treatment of acute infection supplement the replacement. For the phagocyte defects, antibacterial and antifungal prophylaxis is central — co-trimoxazole and an agent such as itraconazole in chronic granulomatous disease — with early surgical drainage of abscesses and a low threshold for targeted anti-mould therapy. For complement deficiency, meningococcal vaccination against the relevant serogroups and a standby oral antibiotic for the family to take at the first sign of fever are the core measures. [7] [8]
For the combined immunodeficiencies including SCID, the only curative therapy is haematopoietic stem cell transplantation, increasingly complemented by gene therapy for specific conditions such as adenosine-deaminase deficiency and X-linked SCID. The timing is the load-bearing point: transplantation performed before the age of three to four months, before the child has acquired serious infection, is associated with markedly higher survival than transplantation after infection has occurred, which is the entire rationale for newborn TREC screening. For the immune-dysregulation disorders, immunosuppression, biological therapy such as mTOR inhibitors in autoimmune lymphoproliferative syndrome, and haematopoietic stem cell transplantation for the most severe forms are the modalities. Across all categories, vaccination strategy is critical: killed and subunit vaccines are given, but live vaccines — including bacille Calmette–Guérin, rotavirus, measles-mumps-rubella and varicella — are withheld until immunology has cleared the child, because they can cause disseminated disease in a profoundly immunodeficient host. [9] [11]
Specific Subtypes & Scenarios
The SCID infant — the time-critical prototype. An infant presents with failure to thrive, persistent oral thrush, recurrent or persistent chest infection, and a low lymphocyte count for age. The scenario is SCID until proven otherwise, and the response is same-day immunology referral, flow cytometry, TREC and genetic testing, prophylactic co-trimoxazole, withholding of live vaccines, irradiated and cytomegalovirus-safe blood products, and a pathway to haematopoietic stem cell transplantation before infection. Newborn screening by the TREC assay identifies these infants before they become symptomatic, and the longitudinal linkage data show a clear survival benefit for transplantation performed after a screen-detected, infection-free diagnosis. [6] [11]
The antibody-deficient child — the recurrent sinopulmonary pattern. A school-age child presents with a history of recurrent otitis media, sinusitis and pneumonia, with two hospital admissions and emerging bronchiectasis. Serum immunoglobulins show low IgG and IgA, and a vaccine-antibody response is poor. The scenario is a predominantly antibody deficiency — common variable immunodeficiency or, in a younger boy with absent tonsils and absent B-cells, X-linked agammaglobulinaemia — and the management is immunoglobulin replacement therapy, antibiotic prophylaxis and structured respiratory surveillance to prevent bronchiectasis. [7]
The phagocyte-deficient child — the abscess and granuloma pattern. A toddler presents with recurrent deep abscesses of the skin, lymph nodes and liver, osteomyelitis, and granulomatous inflammation, growing Staphylococcus aureus, Serratia or Aspergillus. The dihydrorhodamine test is abnormal, confirming chronic granulomatous disease. The management is co-trimoxazole and itraconazole prophylaxis, early drainage, anti-mould therapy for suspected Aspergillus, and consideration of interferon-gamma and haematopoietic stem cell transplantation in selected cases. [7] [8]
The complement-deficient adolescent — the Neisseria pattern. An adolescent presents with meningococcaemia or recurrent invasive Neisseria disease. The total haemolytic complement CH50 is low, and individual component testing identifies a terminal-pathway deficiency. The management is meningococcal vaccination against serogroups ACWY and B, a standby oral antibiotic for the family to take at the first sign of fever, and clear safety-netting advice. The same adolescent should be screened for autoimmune thyroid and connective-tissue disease, which are associated with early complement-component deficiency. [8]
Complications & Pitfalls
The central pitfall, and the one that causes preventable death, is diagnostic delay — the treatment of infection after infection as 'just another viral illness' in a child who in fact has a combined immunodeficiency. The reasons are familiar: the high background rate of normal childhood infection that desensitises the clinician, the failure to apply the warning-sign thresholds, the misinterpretation of a lymphocyte count that is 'normal' at an adult threshold but lymphopenic for age, and the absence of a family history that is never elicited. The corrective is the disciplined application of the warning signs and the tiered workup, with a low threshold for the screening immunoglobulins and full blood count in any child whose pattern crosses the line. [1] [5]
The second pitfall is the live vaccine given to a child with unrecognised immunodeficiency. Bacille Calmette–Guérin can disseminate in SCID or chronic granulomatous disease; rotavirus vaccine can cause persistent infection and diarrhoea in SCID; measles-mumps-rubella and varicella can cause severe disease in profoundly immunodeficient hosts. The rule is that any child under investigation for a significant immunodeficiency does not receive live vaccines until immunology has formally cleared the immune function. The third pitfall is the failure to exclude secondary immunodeficiency — most importantly HIV — before settling on a primary cause, which denies the child the correct and often simpler management of the underlying condition. [5] [9]
Prognosis & Disposition
The prognosis of primary immunodeficiency is determined almost entirely by the diagnosis and the timing of intervention. For severe combined immunodeficiency, the prognosis transformed with newborn TREC screening and transplantation before infection: the longitudinal data linking screen-detected SCID to survival after haematopoietic cell transplantation demonstrate a clear and durable benefit, and a child transplanted early and infection-free can expect near-normal survival. For the predominantly antibody deficiencies, immunoglobulin replacement prevents the bronchiectasis and end-organ damage that once defined the natural history, and the prognosis is one of near-normal life expectancy with lifelong therapy and surveillance. [6] [11]
For the phagocyte and complement deficiencies, prognosis depends on adherence to prophylaxis and the speed of treatment of breakthrough infection. For the immune-dysregulation disorders, prognosis depends on the specific condition and on the response to immunosuppression or transplantation. The disposition for every child is immunology-led, multidisciplinary care: a written care plan, a named immunologist, structured infection-prevention education for the family and school, genetic counselling for the parents and extended family, and a planned transition to adult immunology services. The paediatrician's role is to make the diagnosis early, initiate the tiered workup, refer correctly and co-manage with the family — because inborn errors of immunity are lifelong conditions that change the whole family's future. [8] [10]
Special Populations
The newborn with an abnormal screening result. A low or absent TREC result on the newborn bloodspot screen demands urgent recall, confirmation with flow cytometry, prophylactic co-trimoxazole, withholding of live vaccines including rotavirus, irradiated and cytomegalovirus-safe blood products, and a defined pathway to transplantation. The family requires careful, accurate communication: the result indicates T-cell lymphopenia, which has a differential beyond SCID, and the confirmatory testing will define the diagnosis. The newborn screening programmes across the United States and an increasing number of other countries have established the TREC assay as the standard of care, and the candidate should know the operational pathway from screen to transplant. [6] [12]
Indigenous, migrant and remote-area children. Children in remote and indigenous communities face the dual challenge of high background infection exposure (which can mask an immunodeficiency) and reduced access to the staged immunology workup and to immunoglobulin services. The stance is a heightened index of suspicion, use of telehealth and retrieval to bridge the distance, and explicit attention to the social determinants that drive recurrent infection independent of an immune defect. Migrant and refugee children may carry untreated or partially treated infection, incomplete vaccination records that confound the vaccine-antibody response, and consanguinity that increases autosomal-recessive risk — each requiring tailored assessment. [10]
Evidence, Guidelines & Regional Differences
The evidentiary base for primary immunodeficiency recognition and diagnosis has matured rapidly. The International Union of Immunological Societies expert-committee classification — updated in 2019 and again in 2022 — provides the canonical taxonomy of inborn errors of immunity across ten categories, and is the reference point for any fellowship answer on classification. The European Society for Immunodeficiencies multi-stage diagnostic protocol, published by de Vries and colleagues in 2012, operationalises the tiered workup for the non-immunologist and remains the standard framework. The ESID registry working definitions, published by Seidel and colleagues in 2019, provide the clinical diagnostic criteria that synthesise laboratory and clinical findings into defensible diagnoses. [2] [5] [8]
The warning-signs evidence is anchored by the United Kingdom prospective study of Subbarayan and colleagues in 2011, which tested the Jeffrey Modell signs in a paediatric immunology population and established their discriminant value, while acknowledging that no single sign is perfectly sensitive or specific. The antibody-deficiency review by Fried and Bonilla in Clinical Microbiology Reviews remains the authoritative pathophysiology and management reference for the humoral arm. The epidemiological picture is drawn from the United States prevalence study of Kobrynski and colleagues, covering 2001 to 2007, and the registry data of the European network. [1] [7] [10]
The newborn screening evidence is the region in which practice has changed fastest and where regional differences matter most. The eleven-programme United States cohort of Kwan and colleagues in 2014 established the diagnostic yield of the TREC assay, and the California cohort of Amatuni and colleagues extended the experience to 2017. The longitudinal data of Thakar and colleagues in the Lancet in 2023, linking newborn screening to survival after transplantation over nearly four decades, is the definitive evidence that early, screen-detected, infection-free transplantation transforms the prognosis of SCID. Regional differences are real: newborn SCID screening is now standard across the United States, Canada and much of Europe, but availability and funding vary elsewhere, and the candidate should know whether their own jurisdiction screens and what the TREC pathway is locally. [6] [11] [12]
Exam Pearls
- Primary immunodeficiency — now framed as inborn errors of immunity — is suspected when infection becomes recurrent, severe, persistent or unusual; the bedside tool is the Jeffrey Modell ten warning signs. [1]
- The IUIS 2022 classification groups inborn errors of immunity into ten categories; knowing the shape of the categories anchors any answer on classification. [3]
- The diagnostic approach is tiered: tier one is FBC with differential and serum immunoglobulins; tier two adds lymphocyte subsets and vaccine-antibody response; tier three adds neutrophil-function testing, complement, TREC/KREC and genetic sequencing. [5]
- Each immune arm has a signature infection: T-cell or combined — opportunism (Pneumocystis, CMV, Candida); antibody — recurrent sinopulmonary with encapsulated bacteria; phagocyte — catalase-positive abscesses (S. aureus, Burkholderia, Aspergillus); complement — Neisseria. [7]
- Lymphopenia and failure to thrive in an infant is SCID until proven otherwise — same-day immunology referral, flow cytometry and a pathway to transplant. [9]
- Newborn SCID screening uses the T-cell receptor excision circle (TREC) assay; low or absent TRECs flag T-cell lymphopenia before infection occurs. [6]
- Transplantation before three to four months, before infection, transforms survival in SCID — the whole purpose of newborn screening. [11]
- Always exclude secondary immunodeficiency — especially HIV — before settling on a primary cause. [5]
- Withhold live vaccines (BCG, rotavirus, MMR, varicella) in any child under investigation for significant immunodeficiency, until immunology clears immune function. [9]
- Antibody deficiency is managed with immunoglobulin replacement; phagocyte defects with co-trimoxazole ± itraconazole prophylaxis; complement deficiency with meningococcal vaccination and standby antibiotics. [7] [8]
- A normal total lymphocyte count does not exclude a T-cell defect if subsets are abnormal, and a normal immunoglobulin level does not exclude a functional antibody deficiency — the vaccine-antibody response is the functional test. [8]
- Remember the age-specific lymphocyte reference range: infants normally have high counts, so an apparently 'normal' count may be lymphopenic for age and the reason SCID is missed. [12]
References
- [1]Subbarayan A; Colarusso G; Hughes SM; Gennery AR; Slatter M; Cant AJ; Barge D; Flood T; Abinun M; Hambleton S Clinical features that identify children with primary immunodeficiency diseases. Pediatrics, 2011.PMID 21482601
- [2]Tangye SG; Al-Herz W; Bousfiha A; Chatila T; Cunningham-Rundles C; Etzioni A; et al Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol, 2022.PMID 35748970
- [3]Bousfiha A; Moundir A; Tangye SG; Picard C; Ochs HD; Casanova JL; et al The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol, 2022.PMID 36198931
- [4]Bousfiha A; Jeddane L; Picard C; Ailal F; Bobby Gaspar H; Al-Herz W; et al Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol, 2020.PMID 32048120
- [5]de Vries E; European Society for Immunodeficiencies (ESID) members Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol, 2012.PMID 22132890
- [6]Kwan A; Abraham RS; Currier R; Brower A; Andruszewski K; Fuller TM; et al Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA, 2014.PMID 25138334
- [7]Fried AJ; Bonilla FA Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Rev, 2009.PMID 19597006
- [8]Seidel MG; Kindle G; Gathmann B; Quinti I; Buckland M; van Montfrans J; et al The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract, 2019.PMID 30776527
- [9]Kwan A; Puck JM History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol, 2015.PMID 25937517
- [10]Kobrynski L; Powell RW; Bowen S Prevalence and morbidity of primary immunodeficiency diseases, United States 2001-2007. J Clin Immunol, 2014.PMID 25257253
- [11]Thakar MS; Logan BR; Puck JM; Pai SY; Notarangelo LD; Satter LF; et al Measuring the effect of newborn screening on survival after haematopoietic cell transplantation for severe combined immunodeficiency: a 36-year longitudinal data linkage study. Lancet, 2023.PMID 37352885
- [12]Amatuni GS; Currier RJ; Church JA; Lin P; McGhee SA; Murrell K; et al Newborn Screening for Severe Combined Immunodeficiency and T-cell Lymphopenia in California, 2010-2017. Pediatrics, 2019.PMID 30683812