Paeds · allergy-and-immunology
Secondary immunodeficiency
Also known as Acquired immunodeficiency · Secondary immune deficiency · Secondary hypogammaglobulinaemia · Iatrogenic immunodeficiency · HIV-related immunodeficiency in children
A fellowship approach to secondary (acquired) immunodeficiency in children: recognise that it is commoner than primary inborn errors of immunity, sort it into six mechanism-based categories (infection, malignancy and its treatment, drugs and biologics, protein loss, nutritional, splenic and metabolic), link each cause to the immune arm it disables and the infection pattern that follows, then apply the five-step management ladder — treat the cause, give antimicrobial prophylaxis, replace what is missing, vaccinate safely, and surveil — reserving immunoglobulin replacement for proven infection-associated hypogammaglobulinaemia rather than a single low number.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A four-year-old girl on long-term high-dose corticosteroids for an autoimmune disease presents with a dry cough, hypoxia, and bilateral interstitial infiltrates. The same "immunocompromised" label could be written on the chart of a neutropenic oncology child with staphylococcal bacteraemia, a boy with nephrotic syndrome and pneumococcal peritonitis, and a refugee infant with severe malnutrition and Gram-negative sepsis — yet each is failing a different immune arm, will be killed by a different organism, and needs a different defence. Holding the cause-to-arm-to-organism chain in your head is what turns "immunocompromised" from a vague warning into a plan, and that chain is the whole of this topic. [1] [4]
C.A.U.S.E.S.
Overview & Definition
A secondary immunodeficiency is a state in which an otherwise normal immune system is disabled by an acquired external factor, leaving the child vulnerable to infection. The defect is not inherited; it is imposed by an infection, a malignancy and its treatment, a drug, the loss of immune proteins, malnutrition, or the loss of a lymphoid organ. Because the cause is external, much of the damage is reversible if the cause is treated — and that reversibility is the feature that most distinguishes secondary from primary immunodeficiency in practice. [1] [2]
The reason secondary immunodeficiency dominates clinical paediatrics is simple arithmetic. The conditions that produce it — oncology treatment, transplant immunosuppression, biologics, nephrotic syndrome, severe malnutrition, and HIV — are each individually far commoner than even the most frequent primary antibody defect. A general paediatrician will meet secondary immune failure many times a year and a primary inborn error perhaps a few times in a career. Defaulting to the question "what is disabling this child's immunity?" before reaching for a primary-disease work-up is the discipline of the topic. [2] [4]
The framework that organises every secondary cause is mechanism-based: sort by which arm of the immune system the cause disables. A cause that depletes CD4 T cells (HIV) produces opportunistic infection; one that wastes IgG (nephrotic syndrome) produces encapsulated-bacterial infection; one that causes neutropenia (chemotherapy) produces Gram-negative and staphylococcal bacteraemia. The mechanism predicts the organism, and the organism predicts both the empiric treatment and the prophylaxis you put in place afterwards. [1] [10]
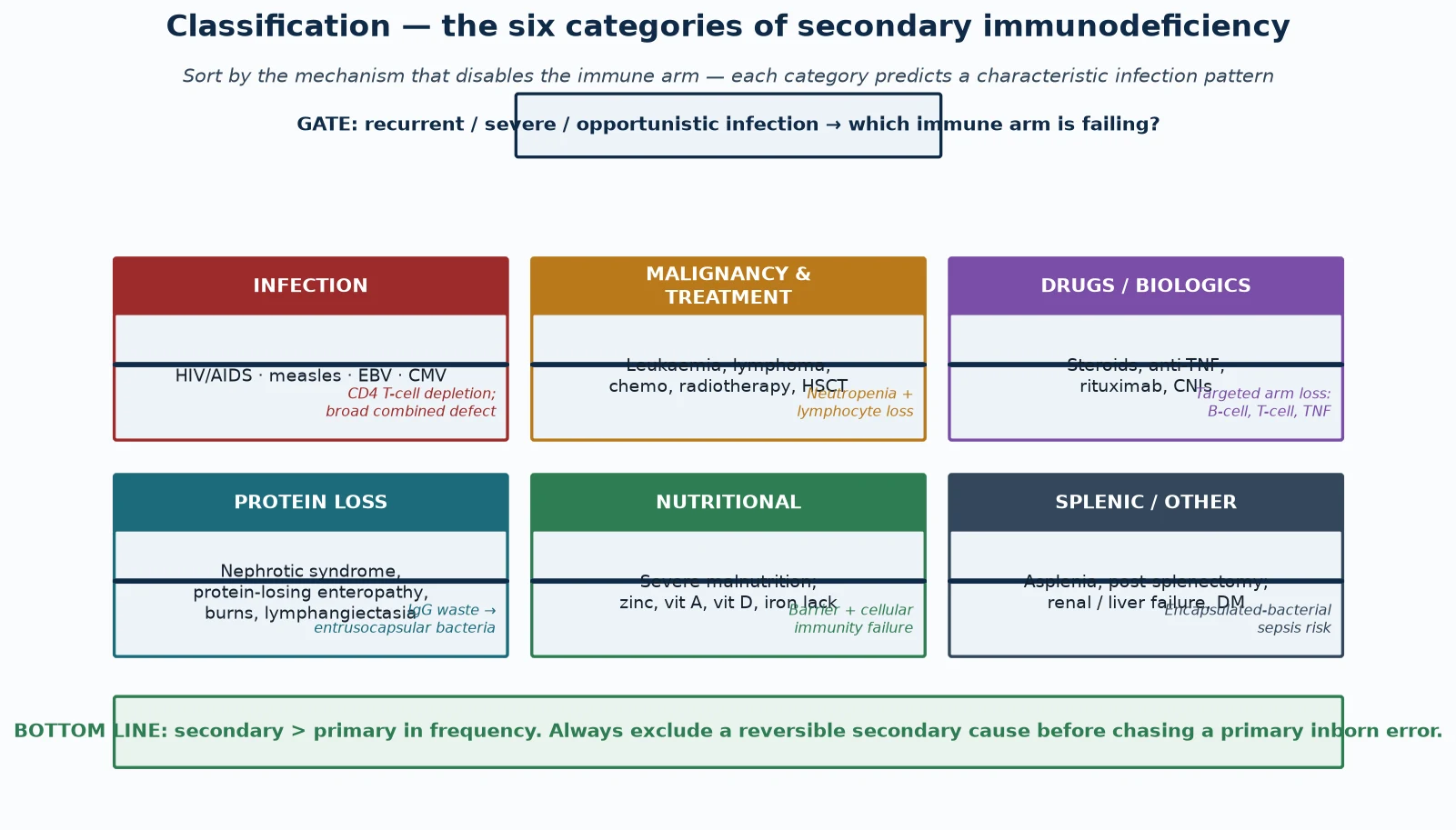
Classification
Sort secondary immunodeficiency into six mechanism-based categories, because the category predicts the disabled arm, the infecting organism, and the prophylaxis in one move. A fellowship answer that merely lists causes without linking each to its mechanism reads as a catalogue and fails to teach. [1] [2]
Infection-driven immunodeficiency is dominated by HIV, which progressively depletes CD4 T cells and produces the widest infection susceptibility of any secondary cause — Pneumocystis, candidiasis, cytomegalovirus, mycobacteria, and herpesviruses. Measles, Epstein-Barr virus, and cytomegalovirus can also produce transient immune suppression in children. This category demands antiviral treatment of the cause (antiretroviral therapy for HIV) as well as prophylaxis against the predicted opportunists. [1]
Malignancy and its treatment disable immunity through both the disease and its therapy. Leukaemia and lymphoma infiltrate and displace normal haematopoiesis, while chemotherapy and radiotherapy cause profound neutropenia, mucosal barrier injury, and lymphocyte loss. Haematopoietic stem cell transplant adds a prolonged period of T-cell deficiency and graft-versus-host risk. The crossover between primary immunodeficiency and malignancy is real, because some inborn errors themselves predispose to haematological cancer. [4] [6]
Drugs and biologics disable a chosen immune arm by design. Corticosteroids suppress T-cell activation, cytokine production, and macrophage function; calcineurin inhibitors block T-cell signalling; anti-TNF agents impair granuloma integrity; and rituximab depletes CD20-positive B cells and can cause late, persistent hypogammaglobulinaemia. Each carries a predictable infection signature, which is why the medication list is part of the diagnosis. [10] [8]
Protein loss wastes immunoglobulin and complement. Nephrotic syndrome loses IgG and alternative-pathway complement factors (notably factor B and properdin) in the urine, protein-losing enteropathy and intestinal lymphangiectasia lose protein across the gut, and extensive burns lose immunoglobulin and complement through the wound. The result is susceptibility to encapsulated bacteria, classically pneumococcal peritonitis in nephrotic relapse. [8] [3]
Nutritional immunodeficiency is the commonest secondary cause globally. Severe malnutrition causes thymic atrophy, barrier breakdown, and failure of cell-mediated immunity, and specific micronutrient deficiencies — zinc, vitamin A, vitamin D, and iron — each impair defined immune functions. The systematic-review evidence shows measurable, broad immune dysfunction in malnourished children, and refeeding restores much of it. [5] [2]
Splenic and metabolic causes form the sixth group. Asplenia and hyposplenism — whether surgical, congenital, or functional (sickle-cell disease) — abolish opsonisation and clearance of encapsulated bacteria, producing the risk of overwhelming post-splenectomy infection. Chronic renal and liver failure and diabetes add combined immune impairment through uraemia, hypogammaglobulinaemia, and neutrophil dysfunction. [9] [1]
Epidemiology & Risk Factors
Secondary immunodeficiency is the commonest form of immune failure encountered in paediatric practice, and its epidemiology tracks the prevalence of its causes. The improvement in survival of childhood cancer, the expansion of transplant and biologic therapy, the persistence of HIV in endemic regions, and the global burden of severe malnutrition together make acquired immune failure a routine rather than an exotic problem. [2] [4]
The highest-risk paediatric populations are identifiable from the clinical context. Oncology patients during and after chemotherapy, recipients of solid-organ or haematopoietic stem cell transplants, children on chronic high-dose corticosteroids or biologics such as rituximab and anti-TNF agents, children with nephrotic-range proteinuria or protein-losing enteropathy, severely malnourished infants, HIV-exposed and infected children, and asplenic or functionally hyposplenic children (notably sickle-cell disease) each carry a predictable, mechanism-specific infection risk. [1] [3]
Severe malnutrition and HIV dominate the global burden. In low- and middle-income settings, malnutrition is the leading cause of acquired immune failure in young children, and the immune dysfunction it produces — measurable at the cellular level — accounts for a large fraction of infection-related child mortality worldwide. HIV-related immunodeficiency remains a major cause where vertical transmission programmes are incomplete. [5] [1]
Corticosteroid exposure deserves a specific risk note. The infection risk rises with both dose and duration: a prednisone-equivalent dose of 20 mg daily or more for longer than four weeks meaningfully increases the risk of Pneumocystis, invasive fungal infection, and reactivation of latent organisms. This dose-duration threshold is the practical trigger for Pneumocystis prophylaxis in non-HIV immunocompromised children. [10] [6]
Pathophysiology
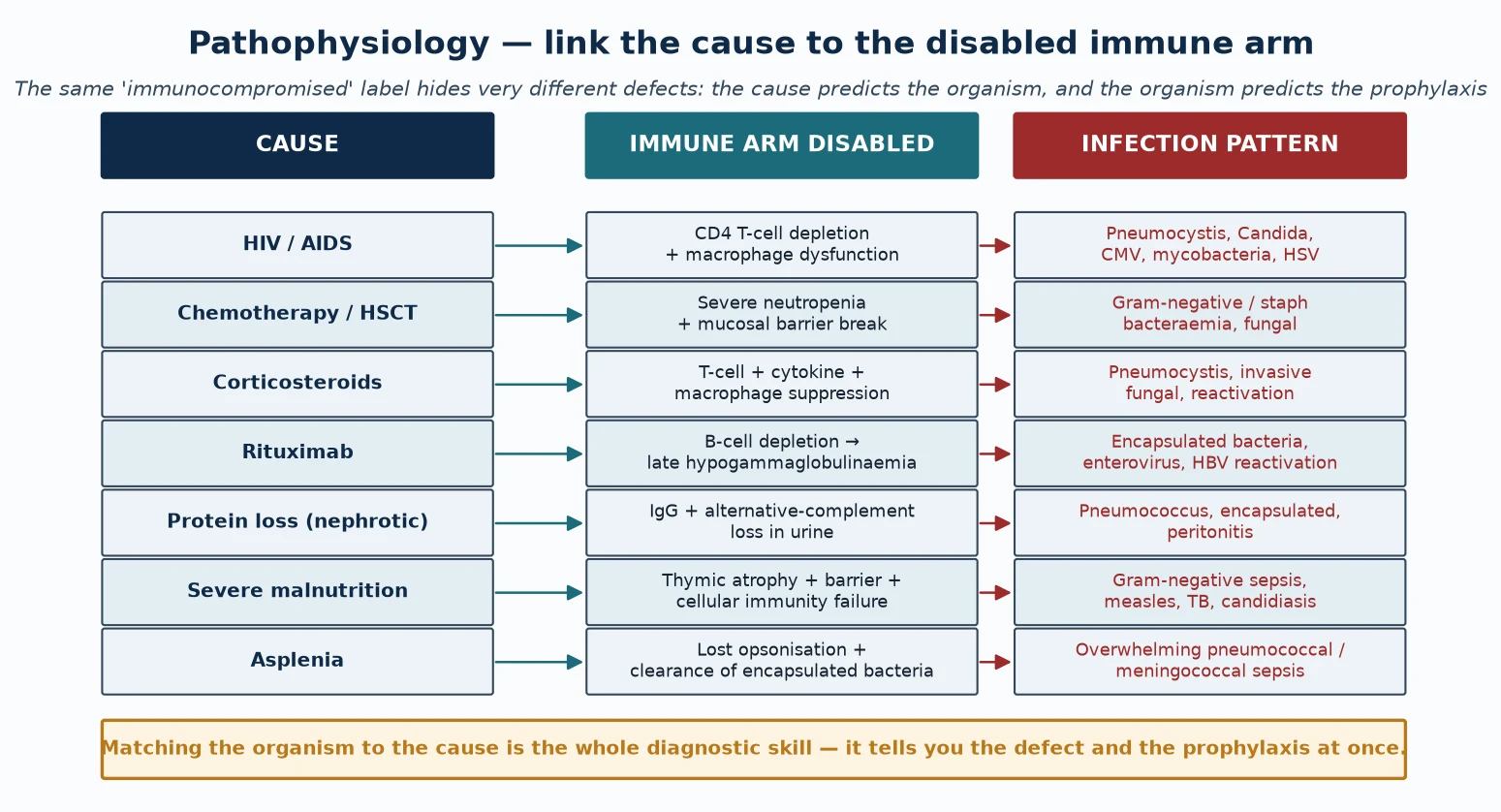
The pathophysiology of secondary immunodeficiency is best held as a three-column map: the cause, the immune arm it disables, and the infection pattern that follows. Reading down that map is how you predict the organism at the bedside and choose the prophylaxis afterwards. The same "immunocompromised" label hides very different defects, and the defect dictates the defence. [1] [4]
HIV infection disables cell-mediated immunity by progressive depletion of CD4-positive T-helper cells and by macrophage dysfunction. As the CD4 count falls, the child loses the ability to contain intracellular organisms, so Pneumocystis pneumonia, oesophageal candidiasis, cytomegalovirus disease, mycobacterial infection, and severe herpesvirus reactivation appear in a predictable sequence. Antiretroviral therapy halts and partially reverses the depletion, but immune reconstitution can itself provoke an inflammatory reaction. [1]
Chemotherapy and haematopoietic stem cell transplant disable immunity chiefly through severe neutropenia and breakdown of the mucosal barrier, compounded later by lymphocyte deficiency and, after transplant, graft-versus-host disease. Neutropenia removes the first-line defence against endogenous bacteria and fungi, so the dominant threats are Gram-negative and staphylococcal bacteraemia and invasive fungal infection, which is why febrile neutropenia is treated as an emergency. [6] [4]
Corticosteroids suppress T-cell activation, cytokine transcription, and macrophage effector function, and they impair the integrity of granulomas. The predictable consequence is susceptibility to Pneumocystis, invasive fungal infection, and reactivation of latent organisms such as tuberculosis, herpesviruses, and strongyloides. The risk is dose- and duration-dependent, which is why a threshold-based prophylaxis rule applies. [10] [6]
Rituximab depletes CD20-positive B cells and, because B-cell reconstitution takes many months and immunoglobulin production recovers slowly, can produce a late and sometimes persistent hypogammaglobulinaemia with recurrent sinopulmonary and enteroviral infection and a risk of hepatitis B reactivation. The drug's use in childhood nephrotic syndrome has made this a routine paediatric scenario rather than a rare one. [8] [3]
Protein loss wastes immunoglobulin and complement. Nephrotic syndrome preferentially loses IgG and alternative-pathway complement factors in the urine because of their relatively small molecular size, leaving the child unable to opsonise and clear encapsulated bacteria — the mechanism behind the classic pneumococcal peritonitis of nephrotic relapse. Protein-losing enteropathy and extensive burns act by analogous loss across a different surface. [8] [3]
Severe malnutrition disables immunity through thymic atrophy (with loss of naive T-cell output), breakdown of skin and mucosal barriers, and failure of both innate and cell-mediated responses, while specific micronutrient deficiencies each impair defined functions — zinc and vitamin A for epithelial integrity and lymphocyte function, iron and vitamin D for phagocyte and antimicrobial defence. Refeeding and micronutrient replacement restore much of the deficit. [5] [2]
Asplenia and hyposplenism abolish the spleen's dual role of opsonising and clearing encapsulated bacteria from the blood. Without it, pneumococci, meningococci, Haemophilus influenzae type b, and, after animal bites, Capnocytophaga can produce overwhelming sepsis within hours. Functional asplenia in sickle-cell disease behaves identically, which is why prophylaxis and vaccination apply to both surgical and functional cases. [9]
Clinical Presentation
The clinical fingerprint of secondary immunodeficiency is infection that is recurrent, severe, persistent, or caused by an opportunistic or unusual organism, arising in a child whose history already declares a cause. The presentation is rarely a mystery once the cause is sought; the trap is failing to ask. [1] [2]
The infection pattern follows the disabled arm. The HIV-infected child presents with Pneumocystis pneumonia, chronic candidiasis, or recurrent bacterial infection as the CD4 count falls. The neutropenic oncology child presents with febrile neutropenia — fever in the setting of an absolute neutrophil count below 0.5 × 10⁹ per litre — which is a medical emergency. The child on high-dose steroids presents with subacute hypoxic Pneumocystis pneumonia or invasive fungal infection. Each is a different tempo demanding a different response. [10] [6]
The child with protein-loss immunodeficiency presents with the signature infection of nephrotic relapse: spontaneous bacterial peritonitis, classically pneumococcal, alongside oedema and heavy proteinuria. The severely malnourished child presents with Gram-negative sepsis, severe measles, tuberculosis, or persistent candidiasis, often with the stigmata of wasting and oedematous malnutrition. The asplenic child presents with overwhelming encapsulated-bacterial sepsis that can kill within hours of the first fever. [8] [9] [5]
A medication, disease, or exposure in the history is itself the presenting feature that points to the cause. The first question in any "immunocompromised child" is therefore not which test to send but what is disabling the immunity: a drug, a malignancy, a protein-losing state, malnutrition, HIV, or an absent spleen. The history converts the label into a mechanism and the mechanism into a plan. [1] [3]
Differential Diagnosis
The differential diagnosis for recurrent or severe infection in a child is broad, and the discipline of this topic is to think structurally: separate secondary causes, primary inborn errors, and the non-immunological mimics before committing to a label. Most recurrent infection in childhood is physiological or anatomical, not immunological. [2] [1]
Physiological recurrent infection is the commonest explanation. A child in daycare or with school-age siblings can have eight to ten viral upper-respiratory infections a year, each lasting up to two weeks, and still be normal. The discriminating features are severity (deep-tissue or opportunistic infection), persistence (infection that fails to clear), and the organism (encapsulated bacteria or opportunists in an unwell child). [2]
Anatomical and barrier causes mimic immunodeficiency. Recurrent pneumonia in the same lobe raises the question of an inhaled foreign body or a congenital lung malformation; recurrent skin infection may reflect eczema as a portal of entry; chronic suppurative ear disease may reflect eustachian-tube dysfunction. These are excluded by history, examination, and targeted imaging rather than by an immunoglobulin panel. [1]
Primary inborn errors of immunity must be distinguished from secondary causes, because the treatment and counselling differ. The discriminating features are an early, stereotyped infection pattern; a family history of consanguinity, early male death, or known immunodeficiency; and infection that persists or recurs after the secondary cause is removed. A primary defect should not be confirmed until reversible secondary causes have been excluded, because the two can coexist. [1] [4]
A concurrent primary defect in a child with an obvious secondary cause is the trap that costs marks. Rituximab may unmask a previously silent antibody deficiency, and malignancy may arise on the background of an inborn error. Persistent or unusually severe infection that does not fit the secondary mechanism, or that fails to resolve once the cause is treated, should reopen the search for a primary immunodeficiency. [4] [3]
| Pattern | Likely alternative | Key discriminator |
|---|---|---|
| 8-10 viral colds/year, daycare | Normal childhood exposure | Well between episodes, full recovery, normal growth |
| Same-lobe pneumonia repeatedly | Inhaled foreign body or malformation | Localised finding, imaging confirms |
| Opportunistic organism in a well child | Secondary immunodeficiency (drug, HIV, malignancy) | Cause evident on history; mechanism matches organism |
| Persistent infection after cause resolves | Concurrent primary inborn error | Stereotyped pattern, family history, genetic testing |
Clinical & Bedside Assessment
The bedside task is to identify the cause that is disabling immunity and to assess the severity and tempo of the current infection, because both shape the immediate response. Begin with the threat gate: any opportunistic organism, febrile neutropenia, or overwhelming sepsis in a child with a known risk factor is an emergency that earns immediate cultures and treatment while the work-up proceeds. [1] [6]
The history is the discriminating instrument. Establish the full medication and biologic exposure (drug, dose, duration), the oncology and transplant status and phase of treatment, the nutritional state, the splenic status (surgical, congenital, or functional such as sickle-cell disease), and any HIV exposure. Document the number, site, severity, and documented microbiology of infections, and ask directly about Pneumocystis-defining illnesses and overwhelming sepsis. A careful growth and developmental history is essential, because failure to thrive alongside infection changes the threshold to investigate. [10] [2]
The examination carries the physical clues that point to specific causes. Cachexia, muscle wasting, and oedema mark severe malnutrition; oral candidiasis, hairy leucoplakia, or generalised lymphadenopathy point to HIV; tonsillar and nodal atrophy suggest prolonged immunosuppression; localised chest findings suggest pneumonia or Pneumocystis; and signs of shock mark overwhelming sepsis in the asplenic child. Plot growth parameters, because faltering growth with infection is a red flag in every category. [5] [9]
Assess the social and access context at the same visit, because it shapes both the risk and the disposition. A child in a remote community, a refugee or asylum-seeking family, or a household with socioeconomic disadvantage may present late and face barriers to prophylaxis and immunoglobulin access. Documenting these factors shapes the prophylaxis plan, the retrieval pathway, and the transition to shared care, and it is part of the assessment rather than an afterthought. [2] [3]
Investigations
The investigation ladder is cause-directed rather than blanket. Begin with the tests that identify the mechanism, confirm the immunoglobulin defect functionally, and define the infection, then characterise further only as the mechanism demands. A low immunoglobulin number alone is never a diagnosis; it must be paired with a functional assessment and interpreted against the cause. [3] [1]
First-line tests are a full blood count with differential (the absolute neutrophil count defines neutropenia), total immunoglobulins (IgG, IgA, IgM) interpreted against age-adjusted ranges, and a functional vaccine response where a humoral defect is suspected. The vaccine response — specific IgG to pneumococcus before and after polysaccharide challenge — separates a true functional defect from a number, and it identifies the child who may benefit from immunoglobulin replacement. [3] [8]
Cause-specific tests confirm the mechanism. A CD4 count and HIV test are indicated whenever opportunistic infection suggests cell-mediated failure; urine protein and serum albumin define nephrotic-range protein loss; lymphocyte subsets and flow cytometry characterise B-cell depletion after rituximab; and nutritional markers (albumin, zinc, vitamin A and D, iron studies) define micronutrient deficiency. Each test is sent because the history pointed to that arm, not as a screen. [1] [5]
Microbiology and imaging define the opportunistic infection. A chest radiograph and, when Pneumocystis is suspected, a chest computed tomography and induced-sputum or bronchoalveolar-lavage staining establish the organism; blood cultures define bacteraemia; and targeted testing defines cytomegalovirus, mycobacteria, and fungi. The microbiology both guides acute treatment and confirms the mechanism that the history predicted. [6] [7]
Management — Resuscitation
Resuscitation in secondary immunodeficiency means treating the acute infection safely while respecting two cause-specific hazards: live vaccines in a severely immunocompromised child, and the tempo of overwhelming sepsis in neutropenia or asplenia. [1] [6]
Febrile neutropenia is the prototype emergency. A child on chemotherapy with an absolute neutrophil count below 0.5 × 10⁹ per litre and a fever requires blood cultures and empirical broad-spectrum intravenous antibiotics within the hour — typically an antipseudomonal beta-lactam such as piperacillin-tazobactam or ceftazidime — modified by local guidelines and the clinical picture. The principle is that the immune compartment is empty, so a fever is sepsis until proven otherwise. [6] [4]
Overwhelming post-splenectomy infection demands the same urgency. An asplenic or hyposplenic child with fever and signs of sepsis receives empirical intravenous antibiotics active against encapsulated organisms — a third-generation cephalosporin such as ceftriaxone — without waiting for cultures, because the window between first fever and death can be hours. A pre-hospital standby emergency antibiotic supply is part of every asplenic child's action plan. [9]
Acute opportunistic infection is treated empirically as the organism dictates: high-dose cotrimoxazole for Pneumocystis pneumonia, ganciclovir for cytomegalovirus, and antifungal therapy for invasive fungal disease, with cultures and imaging guiding refinement. Oxygen and respiratory support are given as required, and the underlying cause — HIV, steroid excess, transplant immunosuppression — is addressed in parallel because immune recovery is part of the treatment. [1] [7]
The antibody-specific hazard is live vaccination in a child who may be severely immunocompromised. Rotavirus, measles-mumps-rubella, varicella, and BCG vaccines are deferred until immune reconstitution is documented, because they can cause disseminated disease. Household contacts should instead be fully vaccinated to provide indirect protection. [1] [9]
Management — Definitive & Stepwise
Definitive management follows a five-step ladder that applies across every secondary cause: treat the cause, give antimicrobial prophylaxis, replace what is missing, vaccinate safely, and surveil with shared care. Treatment is cause-specific, but protection is general — and holding both halves in view is the fellowship skill. [1] [3]
Step one is to treat the cause. Secondary immunodeficiency is acquired and often reversible, so cause-directed therapy is the foundation: antiretroviral therapy for HIV, remission-induction for the underlying disease in steroid-treated autoimmune conditions, reduction or withdrawal of immunosuppression where feasible, refeeding and micronutrient replacement for malnutrition, and surgical or functional correction of splenic loss where relevant. Treating the cause is the single most effective infection-prevention measure. [1] [5]
Step two is antimicrobial prophylaxis matched to the mechanism. Cotrimoxazole prophylaxis against Pneumocystis is indicated for children on high-dose corticosteroids (prednisone 20 mg daily or more for four weeks or longer), during and after chemotherapy and haematopoietic stem cell transplant until immune reconstitution, and for HIV-infected children based on age-stratified CD4 thresholds; a typical paediatric dose is cotrimoxazole 150 mg per square metre per day orally in divided doses, with the exact regimen set by local guidelines. Penicillin prophylaxis — oral phenoxymethylpenicillin, age-adjusted — is indicated for asplenic and hyposplenic children and during nephrotic relapse. [6] [9]
Step three is to replace what is missing. Immunoglobulin replacement is reserved for the child with proven, infection-associated secondary hypogammaglobulinaemia — after rituximab, in protein-losing states, or in malignancy — and is guided by the infection burden rather than the immunoglobulin number alone. Many secondary defects recover when the cause is treated, so replacement is often temporary; committing a child to lifelong therapy for a reversible number is the harm to avoid. [3] [8]
Step four is to vaccinate safely. Inactivated vaccines are given on schedule, recognising that the response may be attenuated and that post-treatment serology confirms protection for high-risk antigens such as pneumococcus and hepatitis B. Live vaccines are contraindicated during severe immunocompromise and are reintroduced once immune reconstitution is documented; household contacts are fully vaccinated to provide herd protection. [9] [1]
Step five is surveillance and shared care. Monitor the relevant compartment — CD4 count in HIV, neutrophil counts during chemotherapy, immunoglobulin troughs where replacement is given — and perform lung surveillance for children at risk of bronchiectasis. Care is shared with the relevant subspecialty (oncology, immunology, nephrology, infectious diseases), and a structured adolescent transition to adult services preserves continuity. [3] [1]

Specific Subtypes & Scenarios
Each major secondary cause carries a distinctive management point, and a fellowship answer earns depth by handling them individually rather than as a single block. The principles generalise, but the prophylaxis is cause-specific. [1] [2]
HIV-related immunodeficiency is managed with combination antiretroviral therapy, which halts CD4 depletion and restores cell-mediated function, alongside cotrimoxazole prophylaxis against Pneumocystis guided by age-stratified CD4 thresholds. CD4 surveillance and a tailored vaccination schedule — inactivated vaccines on schedule, live vaccines only after immune reconstitution — are part of routine care, and the clinician must recognise immune reconstitution inflammatory syndrome, which can unmask or worsen opportunistic infection shortly after antiretroviral therapy starts. [1]
The oncology and post-HSCT child is managed along the neutropenic-fever pathway during treatment and with cotrimoxazole prophylaxis against Pneumocystis after engraftment, with attention to the prolonged T-cell deficiency and graft-versus-host risk after transplant. Re-immunisation on a defined schedule, careful moulding of prophylaxis to the transplant phase, and shared care with oncology and infectious diseases are the structural elements, because the infection risk changes as immune reconstitution proceeds. [6] [4]
The child on rituximab or with nephrotic-range protein loss is managed by monitoring immunoglobulins and the infection burden and reserving immunoglobulin replacement for proven, infection-associated hypogammaglobulinaemia. Rituximab-induced B-cell depletion can produce late hypogammaglobulinaemia that persists for months to years, so infection presenting long after the last dose is a recognised scenario; in nephrotic relapse, penicillin prophylaxis defends against pneumococcal peritonism while the protein-losing state is brought under control. [8] [3]
The asplenic or severely malnourished child is managed by defending against the signature organism. The asplenic child receives penicillin prophylaxis, pneumococcal, meningococcal, and Haemophilus influenzae type b vaccination, and a sick-day action plan with standby emergency antibiotics; the malnourished child receives cautious refeeding, micronutrient replacement, and prompt treatment of infection, because immune function recovers as nutrition is restored. [9] [5]
Why a single low immunoglobulin does not justify replacement
Many secondary hypogammaglobulinaemias recover when the cause is treated — nephrotic syndrome remits, rituximab B-cells reconstitute, chemotherapy ends. Starting immunoglobulin on a number alone commits a child to a costly, lifelong therapy with infusion-reaction and blood-product risks for a defect that was going to resolve. The AAAAI Work Group frames replacement as a decision driven by proven, recurrent infection alongside the laboratory defect, not by the laboratory value in isolation. [3] [8]
Complications & Pitfalls
The complications of secondary immunodeficiency divide into the consequences of untreated or under-treated immune failure and the consequences of the management itself, and a fellowship answer handles both. The dominant complication in every category is infection — overwhelming sepsis, disseminated opportunistic infection, and structural lung damage. [1] [6]
Overwhelming infection is the acute complication. Febrile neutropenia can progress to septic shock within hours; overwhelming post-splenectomy infection can kill between the first fever and arrival at hospital; and untreated Pneumocystis pneumonia causes respiratory failure in the steroid or transplant child. These are the emergencies that make prophylaxis and sick-day planning non-optional. [6] [9]
Structural lung disease is the chronic complication. Recurrent or undertreated sinopulmonary infection in the child with persistent hypogammaglobulinaemia — after rituximab, in nephrotic syndrome, or in HIV with delayed treatment — can progress to bronchiectasis, which becomes the largest determinant of long-term morbidity. Baseline and surveillance lung imaging belong in the plan for the child at risk. [3] [8]
Immune reconstitution inflammatory syndrome is a complication specific to HIV treatment: starting antiretroviral therapy can provoke an inflammatory response to subclinical opportunistic infection as immunity returns, producing clinical deterioration in the first weeks of treatment. Recognising it prevents the misattribution of worsening to treatment failure. [1]
The pitfalls of management are equally important. The commonest is over-diagnosis: starting lifelong immunoglobulin for a single low number in a child whose defect will recover when the cause is treated. The second is the reverse: giving a live vaccine to a severely immunocompromised child, which can cause disseminated disease. The third is missing a concurrent primary defect because an obvious secondary cause was assumed to explain everything. [3] [4]
Prognosis & Disposition
Prognosis in secondary immunodeficiency is dictated by the underlying cause, the timeliness of cause-directed treatment, and the quality of infection prophylaxis. The central prognostic fact — and the one that distinguishes secondary from primary disease — is that many causes are reversible. [1] [2]
Malnutrition-related immune dysfunction improves substantially with refeeding and micronutrient replacement; nephrotic-range protein loss resolves as the nephrotic syndrome remits; rituximab-related B-cell depletion recovers over months to years; and HIV-related immunodeficiency is transformed by antiretroviral therapy, which restores CD4 counts and prevents the opportunistic infections that once defined the disease. In each, treating the cause is the most effective prognostic intervention. [5] [1]
Causes tied to ongoing treatment carry a prognosis shaped by the treatment duration. Oncology and transplant patients move through phases of risk as immune reconstitution proceeds, and their prognosis improves as the prophylaxis is matched to the phase. Asplenia is permanent, so its prognosis depends entirely on prophylaxis, vaccination, and rapid emergency response, which together reduce but do not abolish the risk of overwhelming sepsis. [6] [9]
Disposition for a general paediatrician is shared care with the subspecialty that owns the cause. The relevant team — oncology, immunology, nephrology, infectious diseases, or haematology — owns the cause-specific treatment and the prophylaxis regimen; the general paediatrician owns acute-infection management, growth and development surveillance, vaccination coordination, and the structured transition to adult care. Early referral at the point of suspicion, rather than after a confirmed label, is the disposition rule. [3] [1]
Special Populations
Secondary immunodeficiency interacts with the child's social and developmental context, and a fellowship answer recognises that the same cause behaves differently across populations. Access, adherence, and late presentation all shape outcome. [2] [3]
Indigenous children, particularly in Australia and New Zealand, face a high background burden of recurrent respiratory infection, crowded housing, and reduced access to specialist services. These factors both raise the threshold for suspecting an immune defect and lower the margin for delay when one exists, and structural lung disease is common, so a confirmed secondary defect must be managed intensively to prevent rapid progression to bronchiectasis. [5] [9]
Migrant, refugee, and asylum-seeking families may have incomplete vaccination records, uncertain HIV exposure, malnutrition on arrival, and barriers to consistent prophylaxis access. A careful reconstruction of the infection and exposure history, confirmation of vaccination and HIV status, and a plan that accounts for mobility and language are essential, with interpreter use and trauma-informed communication as part of the approach. [1] [2]
Socioeconomically disadvantaged families carry a disproportionate burden of recurrent infection and late presentation, and adherence to prophylaxis and immunoglobulin therapy can be the limiting step. Subcutaneous home immunoglobulin therapy, where feasible, improves adherence and reduces travel burden and should be discussed for stable patients; the aim is to fit the therapy to the family's reality rather than the reverse. [3] [8]
Adolescents in transition are a population in their own right. Adherence to prophylaxis and immunoglobulin therapy declines in adolescence, risk-taking rises, and the handover to adult care is a vulnerable point. A structured, documented transition that preserves continuity of the prophylaxis regimen and the surveillance trajectory is the safeguard, and it belongs to the disposition plan. [3] [1]
Evidence, Guidelines & Regional Differences
The evidence base for secondary immunodeficiency in children rests on landmark reviews, consensus guidance for secondary hypogammaglobulinaemia, and prophylaxis guidelines developed for the major at-risk groups. [1] [3]
The Chinen and Shearer review remains the framing reference for secondary immunodeficiency, including HIV, organising the causes and mechanisms that a fellowship answer is built on, and the more recent overviews and Work Group reports refine the framework for contemporary practice. The AAAAI Work Group Report on secondary hypogammaglobulinaemia provides practical, consensus-based guidance for diagnosis and management, including the principle that immunoglobulin replacement is driven by infection burden, not by the laboratory value alone. [1] [2] [3]
Prophylaxis guidelines are the operational evidence. The ECIL guidelines define Pneumocystis prophylaxis for patients with haematological malignancy and stem cell transplant recipients, and the Cochrane review synthesises the evidence for prophylaxis in non-HIV immunocompromised patients. These guidelines differ in threshold and duration between HIV and non-HIV immunocompromised children, and they are adapted locally to product availability and funding. [6] [7]
Cause-specific evidence underpins the management of individual scenarios. The systematic review of the immune system in malnourished children documents the broad cellular dysfunction that refeeding reverses; the literature on rituximab in childhood nephrotic syndrome defines the late hypogammaglobulinaemia that follows B-cell depletion; the asplenia guidance defines prophylaxis and vaccination; and the corticosteroid literature defines the dose-duration threshold for infection risk. [5] [8] [9] [10]
Regional differences are practical rather than scientific. Australia and New Zealand apply the international prophylaxis frameworks with national blood arrangements funding immunoglobulin products, and subcutaneous home therapy is increasingly used for stable patients to improve adherence and reduce travel, particularly in remote communities. Indigenous-health and remote-access considerations intensify the need for early prophylaxis and intensive bronchiectasis prevention. [3] [9]
In Australia and New Zealand, secondary immunodeficiency is managed in shared care between the general paediatrician and the relevant subspecialty, with immunoglobulin products funded through national blood arrangements and subcutaneous home therapy increasingly preferred for stable patients. Asplenic and Indigenous children receive intensified prophylaxis and vaccination, and remote-access considerations favour early referral and home-based therapy where appropriate. [3] [9]
Exam Pearls
A fellowship candidate answering on secondary immunodeficiency should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [1] [3]
Anchor one: secondary immunodeficiency is commoner than primary. Default to the question "what is disabling this child's immunity?" before reaching for a primary-disease work-up, because the cause is usually evident in the history. [2]
Anchor two: the mechanism predicts the organism, and the organism predicts the prophylaxis. HIV depletes CD4 cells and produces Pneumocystis; chemo causes neutropenia and produces Gram-negative bacteraemia; steroids produce Pneumocystis; rituximab and protein loss produce encapsulated-bacterial infection; malnutrition produces Gram-negative sepsis; asplenia produces overwhelming encapsulated-bacterial sepsis. Each has a specific defence. [1] [6]
Anchor three: treat the cause first. Secondary immune failure is often reversible — ART for HIV, refeeding for malnutrition, remission for nephrotic syndrome, withdrawal of immunosuppression — and cause-directed therapy is the most effective infection-prevention measure. [5] [1]
Anchor four: replace immunoglobulin only for proven, infection-associated hypogammaglobulinaemia. A single low number in a reversible cause is not an indication; the Work Group framework is driven by infection burden, not the laboratory value. [3] [8]
Anchor five: defend the child with mechanism-matched prophylaxis and safe vaccination. Cotrimoxazole for Pneumocystis in chemo, steroids, and transplant; penicillin for asplenia and nephrotic relapse; inactivated vaccines on schedule; no live vaccines while severely immunocompromised; and household contacts fully vaccinated. [6] [9]
The three traps to avoid are starting lifelong immunoglobulin for a single low number in a reversible cause, giving a live vaccine to a severely immunocompromised child, and treating febrile neutropenia as anything other than sepsis requiring empirical antibiotics within the hour. Avoid these and the rest of the answer falls into place. [3] [6]
References
- [1]Chinen J, Shearer WT. Secondary immunodeficiencies, including HIV infection. J Allergy Clin Immunol, 2010.PMID 20042227
- [2]Tuano KS, Seth N, Chinen J. Secondary immunodeficiencies: An overview. Ann Allergy Asthma Immunol, 2021.PMID 34481993
- [3]Otani IM, Lehman HK, Jongco AM, Tsao LR, Azar AE, Tarrant TK. Practical guidance for the diagnosis and management of secondary hypogammaglobulinemia: A Work Group Report of the AAAAI Primary Immunodeficiency and Altered Immune Response Committees. J Allergy Clin Immunol, 2022.PMID 35176351
- [4]Ballow M, Sánchez-Ramón S, Walter JE. Secondary Immune Deficiency and Primary Immune Deficiency Crossovers: Hematological Malignancies and Autoimmune Diseases. Front Immunol, 2022.PMID 35924244
- [5]Rytter MJ, Kolte L, Briend A, Friis H, Christensen VB. The immune system in children with malnutrition--a systematic review. PLoS One, 2014.PMID 25153531
- [6]Maertens J, Cesaro S, Maschmeyer G, Einsele H, Donnelly JP, Alanio A. ECIL guidelines for preventing Pneumocystis jirovecii pneumonia in patients with haematological malignancies and stem cell transplant recipients. J Antimicrob Chemother, 2016.PMID 27550992
- [7]Stern A, Green H, Paul M, Vidal L, Leibovici L. Prophylaxis for Pneumocystis pneumonia (PCP) in non-HIV immunocompromised patients. Cochrane Database Syst Rev, 2014.PMID 25269391
- [8]Chan EY, Yap DY, Colucci M, Ma AL, Parekh RS, Tullus K. Use of Rituximab in Childhood Idiopathic Nephrotic Syndrome. Clin J Am Soc Nephrol, 2023.PMID 36456193
- [9]Lee GM. Preventing infections in children and adults with asplenia. Hematology Am Soc Hematol Educ Program, 2020.PMID 33275684
- [10]Youssef J, Novosad SA, Winthrop KL. Infection Risk and Safety of Corticosteroid Use. Rheum Dis Clin North Am, 2016.PMID 26611557