Paeds · allergy-and-immunology
T-cell and combined immunodeficiencies
Also known as Severe combined immunodeficiency · SCID · Combined immunodeficiency · Syndromic immunodeficiency · DiGeorge syndrome immune deficiency · Wiskott-Aldrich syndrome
Fellowship topic on T-cell and combined immunodeficiencies: severe combined immunodeficiency (SCID) as the most severe primary immunodeficiency — a genetic block in T-cell development that is fatal within the first one to two years without curative therapy — and the wider combined and syndromic immunodeficiencies (DiGeorge/22q11.2 deletion, Wiskott-Aldrich, ataxia-telangiectasia, Hyper-IgM/CD40L deficiency, Omenn syndrome, MHC class II deficiency). Why loss of T cells collapses the whole adaptive immune system including antibody production even when B-cell numbers are preserved. SCID classification by immunophenotype (T-B-NK-, T-B-NK+, T-B+NK-, T-B+NK+) and the leading genetic causes (X-linked IL2RG, ADA, RAG1/2, JAK3). The presentation of the SCID infant (severe persistent opportunistic infection — Pneumocystis, CMV, candidiasis, BCGosis — failure to thrive, chronic diarrhoea). Newborn screening by T-cell receptor excision circles (TREC) and confirmation by flow cytometry. The management — protect the infant (no live vaccines, reverse isolation, irradiated blood products, prophylaxis, immunoglobulin replacement), refer urgently to a transplant centre, and cure with haematopoietic stem cell transplantation (best if before 3.5 months and before infection) or gene therapy. The danger of live vaccines, the contraindication to non-irradiated blood, and the role of family screening and genetic counselling.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
SCID in one line — and the rule that saves lives
Overview & Definition
Picture a four-month-old boy, previously well, who has been admitted three times in two months — once for pneumonia that turned out to be Pneumocystis, once for oral thrush that would not clear with the usual cream, and once for chronic diarrhoea and weight loss. His mother mentions that an uncle died as an infant decades ago, "before they could find out what was wrong." His full blood count shows a lymphocyte count that the registrar initially glances past — it is low, but the baby is fighting an infection, so surely it should be high. This child has severe combined immunodeficiency, and that low lymphocyte count, in a sick infant, is the single most important number on the page. [1] [7]
Severe combined immunodeficiency, or SCID, is a group of genetically heterogeneous disorders in which a block in T-cell development produces profound immune failure. It is the most severe of the primary immunodeficiencies. Without curative therapy — haematopoietic stem cell transplantation — the condition is fatal within the first one to two years of life from overwhelming infection, and that single fact is what makes SCID a true paediatric emergency rather than an interesting rare disease. The "combined" in the name captures the central teaching point: even when B-cell numbers are preserved, as they are in the common X-linked form, antibody production collapses because B cells cannot function without T-cell help. [6] [7]
The broader category of combined immunodeficiency (CID) shares the same principle — a T-cell defect that drags down the whole immune system — but with enough residual function that presentation may be later and the course less catastrophic. The syndromic combined immunodeficiencies add a recognisable non-immune phenotype: DiGeorge syndrome (22q11.2 deletion) with its cardiac, palatal, and endocrine features; Wiskott-Aldrich syndrome with eczema and thrombocytopenia; ataxia-telangiectasia with cerebellar degeneration and telangiectasia. The 2024 IUIS classification, led by Poli and colleagues, groups all of these under combined immunodeficiencies and syndromic combined immunodeficiencies, providing the authoritative taxonomy for the field. [4] [6]
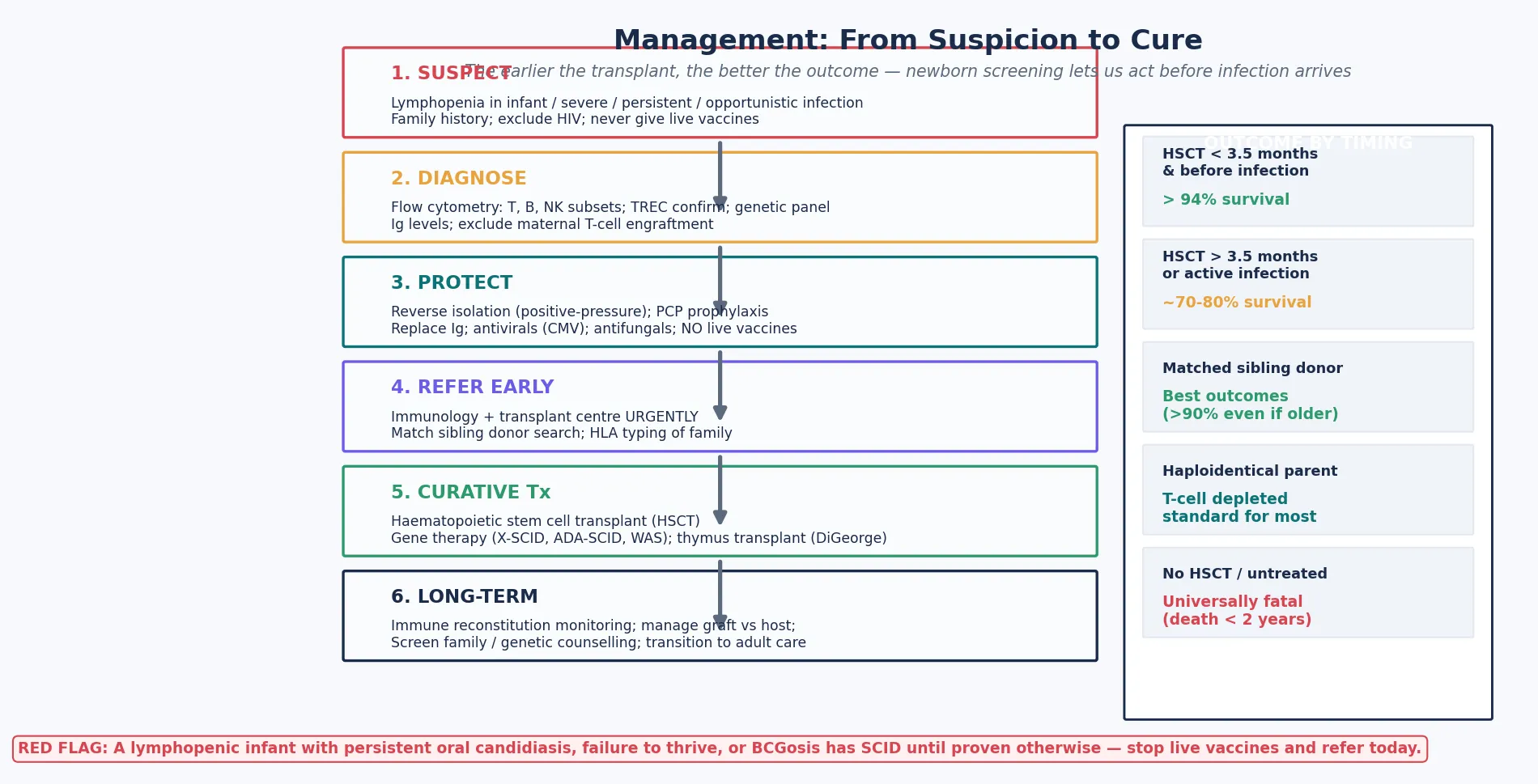
The clinical task has two halves that the examination separates sharply. The first is recognition and protection: suspect SCID from the pattern of infection and the lymphocyte count, confirm with flow cytometry, and protect the infant from iatrogenic harm — live vaccines, non-irradiated blood, and uncontrolled infection. The second is cure: refer urgently to a transplant centre, because the timing of haematopoietic stem cell transplantation, before infection and ideally before 3.5 months of age, is the single most powerful determinant of survival. A candidate who diagnoses SCID but does not understand why it is an emergency, and why the protections matter, fails the question. [1] [2]
Classification
Classification of the combined immunodeficiencies matters because it directs the genetic test, predicts the inheritance and the family counselling, and informs the transplant strategy. The most useful clinical axis is the lymphocyte immunophenotype determined by flow cytometry, because it narrows the genetic differential to a short list before the gene panel returns. [5] [7]
In the T⁻B⁻NK⁻ phenotype, no lymphocytes develop at all. The leading cause is adenosine deaminase (ADA) deficiency, an autosomal recessive disorder in which toxic metabolites accumulate and kill lymphocytes at every stage; it accounts for roughly fifteen to twenty percent of SCID. ADA deficiency is the only SCID form for which enzyme replacement therapy exists — polyethylene glycol-conjugated ADA, or PEG-ADA — and it is one of the established gene-therapy targets. [7]
In the T⁻B⁻NK⁺ phenotype, both T and B cells fail but NK cells are preserved, pointing to a V(D)J recombination defect. The RAG1 and RAG2 genes encode the enzymes that cut T-cell receptor and immunoglobulin genes to create the immune repertoire; their loss prevents receptor assembly. Hypomorphic (partial) RAG mutations produce a different disease — Omenn syndrome, discussed below — in which a few autoreactive T cells leak out and cause a graft-versus-host-like illness. [4] [7]
In the T⁻B⁺NK⁻ phenotype, B cells are present but T cells and NK cells are absent. This is the pattern of the common gamma chain (IL2RG) deficiency, which is X-linked and accounts for roughly forty percent of all SCID — the single commonest form. The same phenotype arises, autosomal recessive, from JAK3 deficiency, because JAK3 is the signalling kinase downstream of the common gamma chain. A boy with this phenotype and a family history of affected maternal uncles has X-linked SCID until the gene test says otherwise. [7]

In the T⁻B⁺NK⁺ phenotype, all lymphocyte lineages except the T-cell block are preserved, pointing to a T-cell-specific signalling or receptor defect — IL-7 receptor alpha deficiency, or defects in the CD3 delta, epsilon, or zeta chains. These are individually rare but collectively important because they are autosomal recessive and may be missed by a family history that is uninformative. The phenotype drives the first genetic guesses, but the definitive answer comes from a targeted gene panel or whole-exome sequencing. [5] [6]
The syndromic combined immunodeficiencies are recognised by their non-immune features. DiGeorge syndrome, from a 22q11.2 deletion, produces thymic hypoplasia and variable T-cell deficiency alongside the cardiac, palatal, and hypocalcaemic features. Wiskott-Aldrich syndrome, X-linked, combines immunodeficiency with eczema and thrombocytopenia with characteristically small platelets. Ataxia-telangiectasia produces progressive cerebellar ataxia, oculocutaneous telangiectasia, and a progressive T-cell decline with malignancy risk. Omenn syndrome is the leaky-SCID face of hypomorphic RAG mutations. The inheritance is worth stating explicitly: X-linked for IL2RG, Wiskott-Aldrich, and CD40L deficiency; autosomal recessive for ADA, RAG1/2, JAK3, ataxia-telangiectasia, and MHC class II deficiency; and autosomal dominant (usually de novo) for most 22q11.2 deletions. [3] [6]
Epidemiology & Risk Factors
Where does SCID sit in paediatric practice, and who is at risk? The incidence, established robustly by the United States TREC newborn screening programs, is approximately one in fifty thousand to one in one hundred thousand live births. The screening data, reported by Kwan and colleagues across eleven programs, captured a higher incidence than pre-screening estimates because it identified atypical and non-classical cases that previously presented only after life-threatening infection. This is the key epidemiological insight: newborn screening did not just detect SCID earlier, it revealed that SCID was more common than we thought. [1]
The risk factors are genetic. Consanguinity increases the rate of autosomal recessive forms, which is why ADA deficiency, RAG1/2 defects, and JAK3 deficiency cluster in consanguineous families and in populations with high rates of cousin marriage. A family history of early infant death from infection — a sibling, a cousin, or an uncle who died before the cause was found — is the single most important historical clue and should prompt the question of whether the family carries a SCID mutation. For X-linked SCID, the mother may be a carrier, and an affected maternal uncle is the classic pedigree. [4] [7]
The cofactors that worsen outcome are iatrogenic and preventable, and the candidate must name them. Administration of live vaccines — BCG, rotavirus, oral polio — to an undiagnosed SCID infant causes vaccine-derived disease: BCGosis, disseminated rotavirus, vaccine-derived poliovirus. Transfusion of non-irradiated blood causes transfusion-associated graft-versus-host disease, which is fatal in SCID. Uncontrolled infection at the time of transplant — CMV, Pneumocystis, adenovirus — is the leading cause of transplant failure and death. Each of these is a preventable harm, and avoiding them is the first job of the clinician who suspects SCID. [9] [10]
Pathophysiology
To understand why SCID is so devastating, you have to understand what T cells do — because the whole disease is a lesson in why losing the conductor wrecks the orchestra. Mature CD4⁺ helper T cells provide the signals that every other limb of adaptive immunity depends on. They express CD40 ligand, which switches B cells from IgM production to IgG, IgA, and IgE — so without T cells, there is no class-switching and no effective antibody. They secrete cytokines that activate macrophages to kill intracellular pathogens and that support CD8⁺ cytotoxic T cells. They orchestrate the response to fungi, viruses, and intracellular bacteria. Remove the T cell, and every section of the immune orchestra loses its cue. [6] [7]
This is why SCID is a combined deficiency even when B-cell numbers are normal. In X-linked SCID, flow cytometry shows B cells — the numbers may even look normal — but those B cells produce only IgM and cannot class-switch or mature their antibody affinity, because the T-cell help they require is absent. The same logic explains why immunoglobulin levels are typically profoundly low across all classes in SCID, and why the child has infections with bacteria, viruses, and fungi alike. The B cell without its T-cell helper is an instrument without a conductor. [7]
The genetic defects map onto the stages of T-cell development, and understanding this map is how you remember the phenotypes. The V(D)J recombination pathway assembles the T-cell receptor in the thymus: RAG1 and RAG2 create the double-strand breaks in the receptor genes, and repair proteins (Artemis, DNA-PKcs) seal them. When RAG1 or RAG2 is defective, no T-cell receptor can be assembled, so no T or B cells mature — the T⁻B⁻NK⁺ phenotype. [7]
The cytokine receptor signalling pathway drives T-cell and NK-cell survival and proliferation. The common gamma chain (IL2RG) is a shared subunit of the receptors for six cytokines — IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 — and its downstream kinase is JAK3. When IL2RG is absent (X-linked) or JAK3 is absent (autosomal recessive), T cells and NK cells cannot receive the signals they need to develop, while B cells are preserved — the T⁻B⁺NK⁻ phenotype. This is the molecular basis of the commonest form of SCID, and it is why X-linked SCID and JAK3 deficiency share the same flow-cytometry signature. [7]
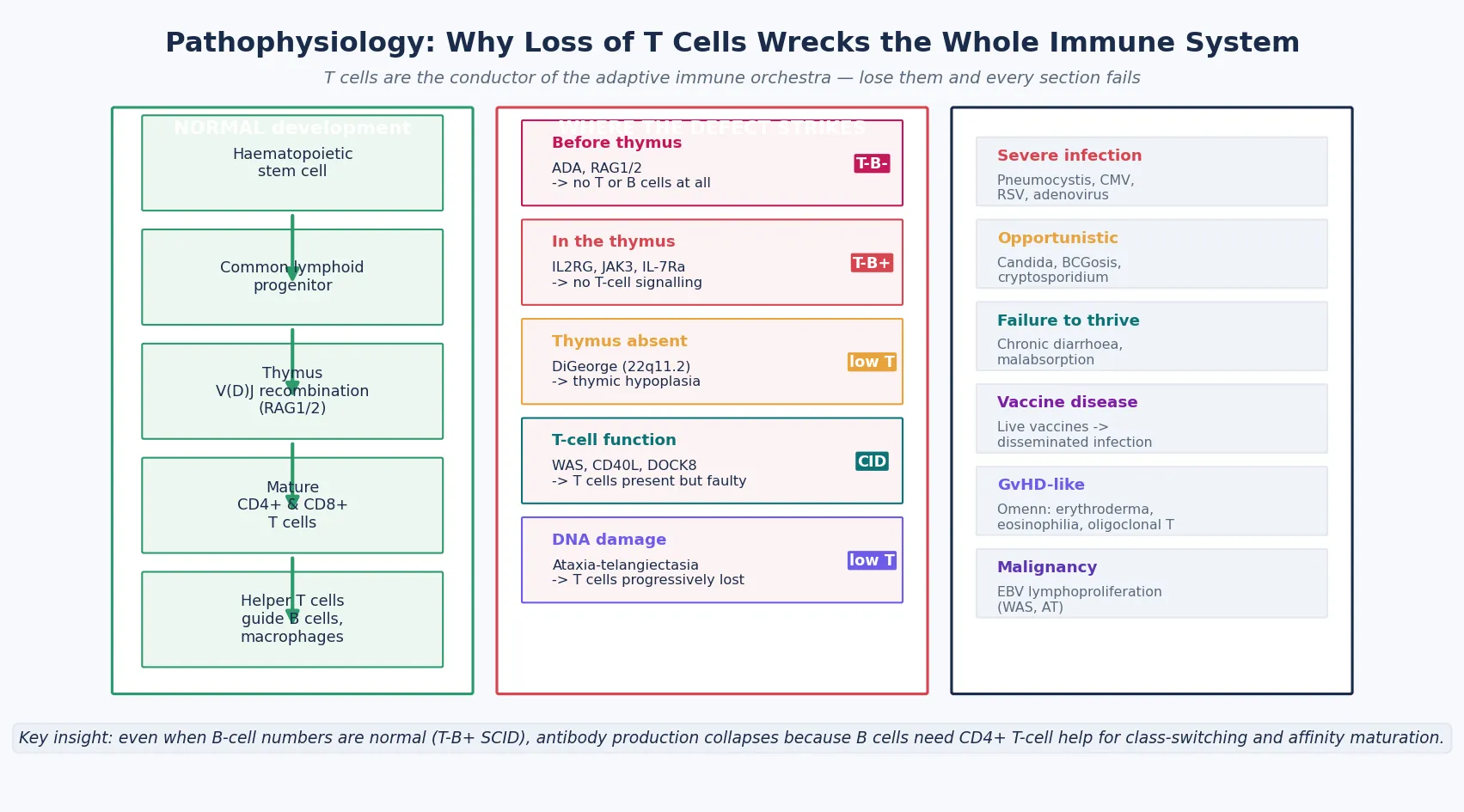
ADA deficiency works differently, by poisoning. Adenosine deaminase normally clears deoxyadenosine; when the enzyme is absent, deoxyadenosine and its phosphorylated derivatives accumulate and are toxic to lymphocytes, which cannot defend against the metabolite. Lymphocytes die at every stage — T, B, and NK alike — producing the T⁻B⁻NK⁻ phenotype. ADA deficiency may also affect non-immune tissues, producing skeletal, hepatic, and neurological features, which is why it is the most clinically varied of the SCID genotypes. [7]
DiGeorge syndrome, by contrast, is a developmental defect rather than a lymphocyte-intrinsic one. The 22q11.2 deletion disrupts the formation of the third and fourth pharyngeal pouches, so the thymus does not develop properly. Without a thymus, T cells cannot undergo their maturation, and the severity of the immune defect tracks with the degree of thymic hypoplasia — from complete athymia (complete DiGeorge, requiring thymus transplantation) to partial forms with near-normal T-cell counts that need only surveillance. The immunodeficiency is one facet of a multisystem syndrome whose cardiac, endocrine, palatal, and neurodevelopmental features dominate the clinical picture. [3] [12]
Omenn syndrome is the disease that teaches you why "leaky" can be as bad as "absent." A hypomorphic RAG mutation allows a few T cells to squeeze through the defective recombination, but they are a narrow, autoreactive, oligoclonal population that attacks the patient's own tissues — a graft-versus-host-like illness of erythroderma, eosinophilia, lymphadenopathy, hepatosplenomegaly, chronic diarrhoea, and a remarkably high IgE. The T-cell count is not low; it is high. The candidate who expects SCID to mean lymphopenia will miss Omenn syndrome unless they know the leaky-SCID phenotype. [4] [7]
Clinical Presentation
The SCID infant presents, almost always, between three and six months of age — the window in which maternally transferred immunoglobulin wanes and the child's own absent immune system is exposed. The infections are severe, persistent, or recurrent, they are caused by organisms that should not trouble a normal infant, and they fail to respond to standard therapy. These three features — severity, opportunism, and treatment failure — are the clinical fingerprint of SCID. [1] [7]
The classic opportunistic infections are the ones to learn by name. Pneumocystis jirovecii pneumonia presents with hypoxia, tachypnoea, and an interstitial infiltrate in a child who has no other reason for it. Cytomegalovirus causes pneumonitis, colitis with chronic diarrhoea, or hepatitis. Refractory oral and diaper candidiasis — thrush that does not clear with topical therapy, spreading to the oesophagus — is one of the earliest signs. Recurrent or severe respiratory viral infections with RSV, adenovirus, or parainfluenza spiral out of control because the child cannot clear the virus. Chronic diarrhoea from enterovirus or rotavirus produces failure to thrive and malabsorption. [7] [10]
Failure to thrive is nearly universal. The weight and length centiles fall away from the birth centiles over weeks to months, driven by chronic infection, diarrhoea, and malabsorption. On examination, the lymph nodes and tonsils are absent or tiny — a critical bedside sign, because it reflects the absent lymphoid development that defines the disease. The experienced clinician who cannot feel cervical nodes or see tonsillar tissue in a sick infant has a powerful clue. [1]
Vaccine-derived disease is a sentinel presentation in countries that immunise at birth. An infant who received the neonatal BCG vaccine develops a non-healing ulcer at the injection site, axillary lymphadenitis, or distant lesions — BCGosis. An infant who received the live rotavirus vaccine develops severe, persistent, or disseminated rotavirus diarrhoea. These are the presentations that bring undiagnosed SCID to attention in the era before newborn screening, and they are preventable if SCID is detected first. [9]
The syndromic forms have their own calling cards. DiGeorge syndrome presents in the neonate with hypocalcaemic seizures (from hypoparathyroidism), a heart murmur from a conotruncal defect (tetralogy of Fallot, interrupted aortic arch, truncus arteriosus), cleft palate, and characteristic facies — hooded eyelids, a small chin, low-set ears. Wiskott-Aldrich syndrome presents with eczema, bruising and bleeding from thrombocytopenia with characteristically small platelets, and recurrent infection. Ataxia-telangiectasia presents after the first year with a progressive cerebellar ataxia that precedes the oculocutaneous telangiectasia. Omenn syndrome presents with the erythroderma, eosinophilia, and lymphadenopathy described above, often misdiagnosed as severe eczema. The candidate who can match the phenotype to the syndrome can direct the genetic test from the bedside. [3] [4]
Differential Diagnosis
The differential diagnosis of the SCID infant is the differential of severe infection with lymphopenia in early infancy. The most important mimic is secondary T-cell deficiency, and the most important secondary cause is vertically acquired HIV. An HIV-exposed infant can present with lymphopenia, failure to thrive, oral candidiasis, and opportunistic infection that looks exactly like SCID. The distinction is the HIV DNA PCR (or RNA viral load), the maternal history, and the flow-cytometry pattern: HIV produces a progressive CD4 decline but preserves total T-cell development and B-cell function, whereas SCID shows an absent T-cell lineage from the start. [7]
Severe malnutrition, immunosuppressive therapy (including prolonged high-dose steroids), and lymphocyte loss through chylothorax, intestinal lymphangiectasia, or thoracic duct disruption can all produce lymphopenia and infection. The history distinguishes these from the congenital immunodeficiency, but the clinician must check the lymphocyte count and act on it. Transient T-cell lymphopenia of prematurity and the benign TREC false-positive — sometimes from limb abnormalities or other non-immune causes of low TRECs — are distinguished from SCID by confirmatory flow cytometry showing a normal or recovering T-cell population. [1] [7]
Isolated antibody deficiency is a critical distinction because the management is completely different. X-linked agammaglobulinaemia (XLA) and common variable immunodeficiency (CVID) present later — after six months, and often after two years for CVID — with recurrent sinopulmonary bacterial infection and normal T-cell counts. They do not cause opportunistic infection or lymphopenia, and they are managed with immunoglobulin replacement, not transplant. The SCID infant is younger, has opportunistic infection, and has an absent or profoundly reduced T-cell population. [5] [6]
Omenn syndrome is mistaken for severe atopic dermatitis, Netherton syndrome, or a primary eczema. The clues that separate Omenn are the lymphadenopathy and hepatosplenomegaly (not features of eczema), the striking eosinophilia and high IgE, the chronic diarrhoea and failure to thrive, and the oligoclonal T-cell population on flow cytometry. Any infant with erythroderma and lymphadenopathy has Omenn syndrome until a hypomorphic RAG mutation is excluded. [4] [7]
Wiskott-Aldrich syndrome is distinguished from immune thrombocytopenia (ITP) by the small platelet size (mean platelet volume is low in WAS, normal or high in ITP), the eczema, the family history of X-linked inheritance, and the combined immunodeficiency on immunological testing. DiGeorge syndrome is distinguished from other causes of hypocalcaemic seizures and congenital heart disease by the 22q11.2 deletion test, which is now a first-line investigation for any infant with a conotruncal cardiac defect and hypocalcaemia. [3] [8]
Clinical & Bedside Assessment
Assess the infant with suspected SCID by looking at the whole child, because the disease is multisystem. Examine the growth chart for the fall-off after the waning of maternal antibody — the weight and length centiles that drift downward over weeks. Feel for the tonsils and lymph nodes, whose absence is a critical sign. Look in the mouth and the diaper for the refractory candidiasis that is one of the earliest clues. Examine the skin for the erythroderma of Omenn or the eczema and bruising of Wiskott-Aldrich. Listen to the chest for the respiratory distress of Pneumocystis or viral pneumonitis. Palpate the abdomen for hepatosplenomegaly. [1] [7]
For the DiGeorge infant, the examination extends to the full syndrome. Auscultate the heart for the murmur of a conotruncal defect. Examine the palate for a cleft or submucous cleft. Look at the facies — the hooded eyelids, small chin, and low-set, posteriorly rotated ears. Test for hypocalcaemia clinically (Chvostek and Trousseau signs) and confirm with biochemistry, because hypocalcaemic seizures may be the presenting event. The immunological assessment is one part of a multisystem evaluation that includes cardiology, endocrinology, and speech and developmental assessment. [3] [8]
The anchoring history is as important as the examination. Ask about the age of onset and the specific organisms, because opportunistic organisms point to SCID. Ask about the vaccine history in detail — did the child receive BCG, and when? Did the child receive the live rotavirus vaccine? Ask about family history, specifically early infant death, consanguinity, and known immunodeficiency. Ask about the maternal HIV status. Document every infection and its organism, because the cumulative infectious burden determines the transplant risk and the urgency of referral. [9] [10]
Inspect the BCG vaccination site specifically. A healthy infant has a small scar or a healed ulcer. An infant with SCID may have a non-healing ulcer, axillary lymphadenitis, or distant cutaneous or systemic lesions — BCGosis. In countries that give neonatal BCG, including much of the world outside the United States, BCGosis is a sentinel presentation of SCID, and the clinician who sees it must act as though SCID is confirmed until flow cytometry says otherwise. [9]
Investigations
The first investigation is the full blood count with attention to the absolute lymphocyte count. Lymphopenia — an absolute lymphocyte count below the age-adjusted normal range — is the bedside clue to SCID, present in the majority of affected infants. Its absence does not exclude SCID, because Omenn syndrome and some T⁻B⁺ forms may have normal or even high T-cell counts, but a low lymphocyte count in a sick infant is a number that must be acted on, not glanced past. [1] [7]
The decisive investigation is flow cytometry for lymphocyte subsets. Measure CD3 (total T cells), CD4 and CD8 (T-cell subsets), CD19 or CD20 (B cells), and CD16 or CD56 (NK cells), and assess naive versus memory T cells and the T-cell receptor repertoire. The pattern determines the immunophenotype — T⁻B⁻NK⁻, T⁻B⁻NK⁺, T⁻B⁺NK⁻, or T⁻B⁺NK⁺ — and narrows the genetic differential to a short list. Flow cytometry also detects maternal T-cell engraftment (mixed chimerism), which can produce a graft-versus-host-like rash and may mask the true phenotype if the maternal T cells are mistaken for the child's own. [7]
Newborn screening by T-cell receptor excision circles (TREC) is the public-health intervention that transformed the prognosis. TRECs are by-products of T-cell receptor rearrangement in the thymus; they are abundant in healthy newborns and low or absent when T-cell development is impaired. Quantification of TRECs from the dried blood spot detects T-cell lymphopenia before symptoms, and a low result prompts confirmatory flow cytometry. The Kwan cohort, spanning eleven United States screening programs, established both the feasibility and the major benefit of universal TREC screening, and it is now standard in many countries. [1]
Genetic testing confirms the molecular diagnosis. A targeted gene panel covering the known SCID and CID genes, or whole-exome sequencing, identifies the mutation and informs the prognosis, the donor choice, the conditioning intensity, and the genetic counselling. Identifying the IL2RG mutation in a boy confirms X-linked SCID and triggers maternal carrier testing; identifying a RAG mutation in an erythrodermic infant confirms Omenn syndrome; identifying an ADA mutation opens the door to enzyme replacement and gene therapy. [6] [7]
Immunoglobulin levels (IgG, IgA, IgM, IgE) are typically profoundly low across all classes in SCID, except IgE, which may be elevated in Omenn syndrome. The low IgG partly reflects waning maternal antibody, but the failure to mount a response confirms the combined defect. Active infection must be investigated thoroughly because the infectious burden before transplant is a major determinant of outcome: request a viral PCR panel (CMV, EBV, adenovirus, enterovirus), Pneumocystis studies if the child is not on prophylaxis, blood and stool cultures, and chest imaging. Each infection found must be treated aggressively before transplant. [10]
Management — Resuscitation
When SCID is suspected or confirmed, the first priority is to protect the infant from iatrogenic harm while the diagnosis is secured and the transplant is arranged. The child is a medical emergency, and the protections are specific, immediate, and non-negotiable. [9] [10]
Stop all live vaccines immediately. BCG, rotavirus, oral polio, and MMR are absolutely contraindicated in SCID because the attenuated organisms that are safe in an immunocompetent child disseminate fatally in a child without T cells. The recommendations of the Medical Advisory Committee of the Immune Deficiency Foundation, published by Shearer and colleagues, are explicit: live bacterial and viral vaccines are withheld from patients with severe T-cell deficiency and their close contacts. If the child has already received a live vaccine before diagnosis, watch for vaccine-derived disease and treat it as a sentinel complication. [9]
Give only irradiated, leucocyte-depleted, CMV-negative blood products. Non-irradiated blood contains viable donor T lymphocytes that can engraft in the immunodeficient recipient and cause transfusion-associated graft-versus-host disease, which is fatal in SCID. Leucodepletion reduces CMV transmission, and CMV-negative products add a further safeguard. This rule applies to every blood product the child receives, from the emergency transfusion to the routine full blood count sample — nothing non-irradiated should touch the child once SCID is on the differential. [10]
Start antimicrobial prophylaxis. Co-trimoxazole provides prophylaxis against Pneumocystis, the classic opportunistic pathogen of SCID. Antifungal prophylaxis (fluconazole or itraconazole) suppresses the refractory candidiasis. Aciclovir or ganciclovir is added if CMV is detected or suspected. These prophylaxes buy time — they do not cure the underlying defect — but they prevent the infections that would worsen the transplant outcome. [10]
Intravenous immunoglobulin (IVIG) or subcutaneous immunoglobulin (SCIG)
Dose
400–600 mg/kg
Give immunoglobulin replacement to provide passive humoral protection. Intravenous or subcutaneous immunoglobulin, typically 400 to 600 mg per kilogram every three to four weeks, supplies the antibodies the child cannot make. Treat active infection aggressively and completely before transplant, because uncontrolled infection is the leading cause of transplant failure and death. Refer urgently to a paediatric immunology and stem cell transplant centre — the timing of transplant, before infection and ideally before 3.5 months of age, is the single most powerful determinant of survival. [2] [10]
Management — Definitive & Stepwise
The curative therapy for SCID is haematopoietic stem cell transplantation, which replaces the defective immune system with healthy stem cells from a donor. The principle is simple — a normal immune system grows back from the donor stem cells — but the execution depends on the donor, the conditioning, and above all the timing. [2] [10]
The donor hierarchy begins with a matched sibling donor, which gives the best outcomes because the donor cells engraft reliably with minimal graft-versus-host disease. When no matched sibling exists — the common situation, given that SCID is rare — the options are a matched unrelated donor from a registry or a haploidentical (half-matched) parent, typically the father. Haploidentical transplants, with T-cell depletion to prevent graft-versus-host disease, have made curative transplantation available to virtually every SCID infant regardless of donor availability. [2] [10]
The timing is the decisive variable, and this is the point the examination tests. Transplant before 3.5 months of age and before the onset of infection achieves survival above 90 to 94 percent. Transplant later in infancy, or with active infection, drops survival to roughly 70 to 80 percent. The Pai cohort, spanning the Primary Immune Deficiency Treatment Consortium transplant era from 2000 to 2009, and the Lankester SCETIDE European cohort, documented these outcomes and established the principle that early, infection-free transplant is the goal. Newborn screening by TREC makes this achievable, because it detects the infant before symptoms and allows transplant in the pre-symptomatic window. [1] [2] [10]
The conditioning regimen — the chemotherapy given before the transplant — is tailored to the donor and the genotype. Matched sibling transplants in young, infection-free infants may use little or no chemotherapy, because the SCID bone marrow is effectively empty and the donor cells engraft without needing space to be created. Unrelated and haploidentical transplants typically use reduced-intensity or myeloablative conditioning — regimens built around treosulfan, fludarabine, or busulfan — to suppress the recipient's residual immunity, create space for the donor cells, and reduce the risk of rejection. The choice balances engraftment against the toxicity of chemotherapy in a small, infected infant. [10]
Gene therapy is an emerging curative option for specific genotypes and is now a real alternative to transplant for some children. For X-linked SCID, ADA-SCID, and Wiskott-Aldrich syndrome, a corrected copy of the defective gene can be inserted into the child's own haematopoietic stem cells using a lentiviral or gamma-retroviral vector, and the corrected cells are returned to the child. Because the cells are the child's own, there is no graft-versus-host disease and no need for a matched donor. Early gamma-retroviral trials were complicated by insertional oncogenesis (leukaemia), but the newer lentiviral vectors and the improved safety profiles have made gene therapy an established option in expert centres for selected genotypes. [6] [10]
For complete DiGeorge syndrome with athymia, the curative option is thymus transplantation — the implantation of cultured postnatal thymus tissue to reconstitute naive T-cell production. The ESID guidelines, led by Kreins and colleagues, set out the selection and management of infants with congenital athymia who are candidates for this procedure. For the majority of 22q11.2 infants with partial forms, the T-cell function is sufficient that no transplant is needed, and the management focuses on prophylaxis, surveillance, and the non-immune comorbidity. [3] [8] [11]
Long-term follow-up after transplant is essential. Monitor immune reconstitution — the recovery of T-cell subsets, the re-emergence of thymic output measured by TREC, and the restoration of antibody production. Manage graft-versus-host disease, which may complicate even matched-sibling transplants. Maintain surveillance for autoimmunity and for malignancy, particularly the EBV-driven lymphoproliferative disease that is a risk in some forms. Plan the transition to adult immunology care for the survivors who reach adolescence and young adulthood. [10]
Specific Subtypes & Scenarios
X-linked SCID (IL2RG) is the commonest single form and the prototype. A boy with the T⁻B⁺NK⁻ phenotype and a family history of affected maternal relatives has X-linked SCID until the gene test confirms it. Maternal carrier testing is available, and prenatal and preimplantation genetic diagnosis allows the family to plan future pregnancies. Gene therapy is now an option alongside transplant. The urgency is unchanged — transplant before infection. [7]
ADA deficiency is the T⁻B⁻NK⁻ form, autosomal recessive, and the most clinically varied because the toxic metabolites affect non-immune tissues. It is the only SCID with enzyme replacement therapy: polyethylene glycol-conjugated ADA (PEG-ADA) restores enzyme activity as a bridge to transplant, and it is one of the established gene-therapy targets. Skeletal anomalies (cupping and flaring of the costochondral junctions), hepatic, and neurological features may coexist. [7]
DiGeorge syndrome (22q11.2 deletion) is the syndromic CID with the widest phenotype. The immune deficiency ranges from complete athymia (complete DiGeorge, requiring thymus transplantation) to partial forms with near-normal T-cell counts that need only surveillance and prophylaxis. The Óskarsdóttir clinical practice recommendations, updated in 2023, set out the multisystem management: cardiology for the conotruncal defect, endocrinology for the hypoparathyroidism, speech and palate assessment, and developmental and psychiatric surveillance. The immunologist's role is to stratify the T-cell deficiency, guide prophylaxis, and identify the minority who need thymus transplantation. [3] [8] [12]
Wiskott-Aldrich syndrome is the X-linked combined immunodeficiency with eczema and thrombocytopenia with characteristically small platelets (low mean platelet volume). The WAS gene product, WASp, regulates the actin cytoskeleton in haematopoietic cells; its loss produces immune dysregulation, autoimmunity, and a high risk of EBV-driven lymphoproliferative disease. Haematopoietic stem cell transplantation is curative, and gene therapy is an emerging option. The bleeding from thrombocytopenia may precede the immune presentation, so any boy with eczema and small-platelet thrombocytopenia has Wiskott-Aldrich until the WAS gene is tested. [6]
Omenn syndrome is the leaky SCID from hypomorphic RAG mutations. The infant presents with erythroderma, eosinophilia, lymphadenopathy, hepatosplenomegaly, chronic diarrhoea, a high IgE, and an oligoclonal T-cell population on flow cytometry. The T-cell count is high, not low, and the disease is often misdiagnosed as severe eczema. Management is the same as classical SCID — protect, treat the skin and infection, and transplant urgently — with the addition that immunosuppression may be needed to control the graft-versus-host-like skin disease before transplant. [4] [7]
Ataxia-telangiectasia is the syndromic CID in which the immune deficiency is progressive rather than congenital. The ATM gene product repairs DNA double-strand breaks; its loss produces cerebellar ataxia (onset after the first year), oculocutaneous telangiectasia (onset later still), progressive T-cell decline, elevated alpha-fetoprotein, and a high risk of malignancy (lymphoma and leukaemia). Management is supportive — immunoglobulin replacement for the antibody deficiency, infection surveillance, and malignancy screening — because haematopoietic stem cell transplantation does not address the neurological disease. The ataxia is usually the first sign, and the telangiectasia and the immune deficiency follow. [5] [6]
Hyper-IgM syndrome from CD40 ligand deficiency is the X-linked defect in immunoglobulin class-switching. The child has low IgG, IgA, and IgE with normal or high IgM, and presents with recurrent bacterial and opportunistic infection — Pneumocystis, and Cryptosporidium causing sclerosing cholangitis. Haematopoietic stem cell transplantation is curative. The name comes from the laboratory pattern — the failed class-switch leaves IgM high while the other classes collapse — and the mechanism is the absence of the T-cell signal (CD40L) that B cells need to switch. [5] [6]
Complications & Pitfalls
The chief avoidable cause of death in SCID is delayed diagnosis. Every day of untreated SCID is a day in which the infant may acquire an irreversible infection that worsens the transplant outcome. Newborn screening by TREC has shifted the diagnosis from the post-infection window — where mortality is high — to the pre-symptomatic window, where survival approaches that of a normal infant. The candidate who understands that SCID is a time-critical emergency, and that the goal is transplant before infection, has the central teaching point of the disease. [1] [2]
The live-vaccine pitfall is preventable and lethal. BCG, rotavirus, and oral polio vaccines, safe in an immunocompetent infant, disseminate fatally in SCID. The rule is absolute: no live vaccines while SCID is being investigated or confirmed, and a live vaccine already given is a reason to watch for and treat vaccine-derived disease. In countries that give neonatal BCG, BCGosis is a sentinel presentation, and the clinician who sees it must act as though SCID is confirmed. [9]
The blood-product pitfall is equally absolute. Non-irradiated blood causes transfusion-associated graft-versus-host disease, which is fatal in SCID because the viable donor T lymphocytes engraft and attack the immunodeficient recipient. Every blood product the child receives must be irradiated and leucocyte-depleted, and CMV-negative products add a further safeguard. This rule applies from the moment SCID is on the differential — not only after it is confirmed. [10]
The lymphopenia pitfall is the failure to act on the absolute lymphocyte count. A low lymphocyte count in a sick infant is abnormal, and the reflexive assumption that lymphopenia in infection is expected (it is, in viral infection, but not in a bacterial or opportunistic infection) leads to missed diagnoses. The candidate who checks the lymphocyte count and acts on it — by ordering flow cytometry — is the one who makes the diagnosis in time. [1]
The maternal T-cell engraftment pitfall produces a graft-versus-host-like rash that is mistaken for a drug reaction or eczema. Maternal T cells cross the placenta and, in a SCID infant who cannot reject them, engraft and attack the skin. Detect it by chimerism studies (mixed lymphocyte populations on flow cytometry) and manage with immunosuppression before transplant. The same pitfall applies to non-irradiated blood — the rash is graft-versus-host disease until proven otherwise. [7] [10]
The transplant complications — graft-versus-host disease, failure of engraftment, and post-transplant lymphoproliferative disorder — determine the long-term outcome and are the focus of post-transplant care. Graft-versus-host disease may complicate even matched-sibling transplants and requires immunosuppression. Failure of engraftment may necessitate a second transplant with intensified conditioning. Post-transplant lymphoproliferative disorder, driven by EBV, is a malignancy risk that mandates viral surveillance and early intervention. [2] [10]
Prognosis & Disposition
Untreated SCID is universally fatal, usually within the first one to two years of life, from overwhelming infection. This is the sentence that defines the urgency of the disease, and the reason every suspected case is a medical emergency. [1] [2]
The transplant outcome data, from the Pai cohort and the Lankester SCETIDE European cohort, establish the principle that timing is everything. Survival above 90 to 94 percent is achievable when transplant occurs before 3.5 months of age and before the onset of infection, falling to roughly 70 to 80 percent when transplant occurs later or with active infection. Matched sibling donors give the best outcomes, with survival above 90 percent even when the transplant is not in the earliest window. Newborn screening by TREC is the tool that makes the early, infection-free transplant achievable, because it detects the infant before symptoms. [1] [2] [10]
For the syndromic CID, the prognosis depends on the specific syndrome. DiGeorge syndrome with partial T-cell function has a good prognosis focused on the non-immune comorbidity — the cardiac, endocrine, and developmental outcomes — while complete athymia requires thymus transplantation. Ataxia-telangiectasia has a progressive neurological and malignant course that haematopoietic stem cell transplantation does not address, with a shortened lifespan driven by the cerebellar degeneration and the malignancy risk. Wiskott-Aldrich syndrome and Hyper-IgM are improved or cured by transplantation, with the caveat that the malignancy risk in WAS must be monitored. [3] [6]
Every infant with confirmed SCID is transferred urgently to a paediatric transplant centre. The family receives genetic counselling, sibling screening, and the offer of prenatal or preimplantation genetic diagnosis for future pregnancies. The long-term survivor enters a transition programme to adult immunology care, with attention to immune reconstitution, autoimmunity, malignancy surveillance, and reproductive and genetic counselling. The arc of the disease — from a near-uniformly fatal condition to one with over 90 percent survival when managed well — is one of the great successes of modern paediatrics, and it rests on the twin pillars of newborn screening and curative transplantation. [1] [10]
Special Populations
In countries with universal TREC screening, the screen-positive infant is referred immediately for confirmatory flow cytometry, and the goal is transplant before infection. This is the population in which the prognosis is transformed, and the clinician's job is to act on the screen without delay. In countries without newborn screening, the infant presents only after life-threatening infection, and the clinician must suspect SCID from the pattern of severe persistent opportunistic infection and lymphopenia and act urgently — the window for infection-free transplant may already be closing. [1]
Families with a known SCID mutation are offered prenatal diagnosis (chorionic villus sampling) or preimplantation genetic diagnosis, and maternal carrier testing for X-linked forms. The power of early intervention on an affected sibling is the difference between near-normal survival and high mortality, so the reproductive counselling is not optional — it is part of the management. The family that has lost one child to SCID, and that is offered prenatal diagnosis for the next pregnancy, is the family that may see their next child transplanted before infection and survive. [4] [6]
For Aboriginal and Torres Strait Islander, Maori, and other Indigenous children, and for refugee, asylum-seeking, and migrant families, the barriers to newborn screening and transplant services must be addressed explicitly. Consanguinity increases the rate of autosomal recessive SCID, which is relevant in some migrant and refugee populations. Culturally safe genetic counselling, with trained interpreters, and equitable access to the transplant pathway are essential — the survival gap that exists when access is uneven is an avoidable injustice. [4]
In rural and remote settings, early recognition of the lymphopenic infant, early retrieval to a transplant centre, and telehealth immunology support are the front line. The family must understand that SCID is an emergency, and that the protections — no live vaccines, irradiated blood, urgent referral — cannot wait for the next routine appointment. The rural clinician who suspects SCID and arranges the flow cytometry and the urgent transfer is the clinician who changes the outcome. [1] [9]
Adolescents with syndromic CID transitioning to adult care — survivors of ataxia-telangiectasia and Wiskott-Aldrich, and transplanted SCID patients entering adulthood — need a structured transition that addresses malignancy surveillance, pulmonary and neurological disease, reproductive and genetic counselling, and the psychosocial adjustment to a chronic immunodeficiency. The transition is not a handover but a planned programme, and its quality determines whether the gains of early transplant are sustained into adult life. [6] [10]
Evidence, Guidelines & Regional Differences
The IUIS classification, updated in 2024 by the Poli committee and previously in the Bousfiha phenotypic classification of 2017, is the authoritative taxonomic reference for the combined immunodeficiencies. The Tangye IUIS interim update of 2021 documented the ever-expanding catalogue of novel inborn errors, and the Notarangelo review of genetically-determined T-cell development defects (2024) set out the mechanistic framework. Together these references define the modern landscape of the field. [4] [5] [6] [7]
The newborn screening and transplant evidence is anchored by two landmark cohorts. The Kwan JAMA 2014 study, spanning eleven United States TREC screening programs, established both the population incidence and the major benefit of pre-symptomatic detection. The Pai NEJM 2014 cohort, from the Primary Immune Deficiency Treatment Consortium, and the Lankester JACI 2022 SCETIDE European cohort, documented the transplant outcomes and the principle that early, infection-free transplant achieves the best survival. These four papers are the evidence base for the modern management of SCID. [1] [2] [10]
For 22q11.2 deletion syndrome, the McDonald-McGinn Nature Reviews Disease Primers article (2015) is the comprehensive reference, the Óskarsdóttir clinical practice recommendations (2023) guide the multisystem management of children, and the van Oers and Sullivan review (2026) sets out the systemic immune effects. The Kreins ESID guidelines (2024) address the management of congenital athymia, including the selection of candidates for thymus transplantation. [3] [8] [11] [12]
The live-vaccine contraindication is set out in the recommendations of the Medical Advisory Committee of the Immune Deficiency Foundation, published by Shearer and colleagues in 2014, which remain the standard reference for vaccination in immunodeficient patients and their close contacts. Regional differences in the vaccine schedule — in particular, whether neonatal BCG and live rotavirus are given — determine which vaccine-derived complications a clinician in a given country is likely to see. [9]
The active controversies include the optimal conditioning regimen intensity for different donor types, the long-term safety of gene therapy (including the insertional oncogenesis events in the early gamma-retroviral trials that led to the development of safer lentiviral vectors), the management of atypical and leaky SCID phenotypes, and equitable access to newborn screening and transplant globally. The candidate should be able to discuss these controversies at viva level, defending the evidence for each position. [6] [10]
Exam Pearls
SCID is the most severe primary immunodeficiency — fatal within one to two years without haematopoietic stem cell transplantation, and a true paediatric emergency. The bedside clue is lymphopenia in a sick infant; the decisive test is flow cytometry. [1] [7]
Classify SCID by the lymphocyte immunophenotype: T⁻B⁻NK⁻ is ADA deficiency; T⁻B⁻NK⁺ is a RAG1/2 recombination defect; T⁻B⁺NK⁻ is X-linked IL2RG (the commonest form) or JAK3; T⁻B⁺NK⁺ is IL-7 receptor alpha or a CD3 component. The phenotype drives the first genetic guesses. [5] [7]
T cells are the conductor of the immune orchestra — even when B-cell numbers are preserved (T⁻B⁺ SCID), antibody production collapses because B cells need CD4⁺ T-cell help for class-switching and affinity maturation. This is why SCID is a combined deficiency even with B cells present. [6] [7]
Newborn screening by TREC detects SCID before symptoms, and transplant before 3.5 months of age and before the onset of infection achieves survival above 90 percent. This is the single most important number in the disease. [1] [2]
Live vaccines are absolutely contraindicated: BCG causes BCGosis, the live rotavirus vaccine causes disseminated disease, and oral polio is dangerous. All blood products must be irradiated and leucocyte-depleted to prevent transfusion-associated graft-versus-host disease and CMV transmission. [9] [10]
DiGeorge syndrome (22q11.2 deletion) is cardiac plus palate plus hypocalcaemia plus thymic hypoplasia; complete athymia needs thymus transplantation, but the majority have partial forms needing only prophylaxis and surveillance. [3] [8]
Omenn syndrome is leaky SCID from a hypomorphic RAG mutation: erythroderma, eosinophilia, lymphadenopathy, high IgE, and an oligoclonal T-cell population — the T-cell count is high, not low, and the disease is often misdiagnosed as severe eczema. [4] [7]
Wiskott-Aldrich syndrome is eczema plus thrombocytopenia with small platelets (low mean platelet volume) plus combined immunodeficiency, X-linked, from the WAS gene. Ataxia-telangiectasia is cerebellar ataxia plus telangiectasia plus sinopulmonary infection plus elevated alpha-fetoprotein plus malignancy risk, and haematopoietic stem cell transplantation does not fix the neurology. ADA deficiency is the only SCID with enzyme replacement (PEG-ADA) as a bridge to transplant, and it is an established gene-therapy target. [5] [6] [7]
References
- [1]Kwan A; Abraham RS; Currier R; Brower A; Andruszewski K; Abbott JK; Baker M; Ballow M Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA, 2014.PMID 25138334
- [2]Pai SY; Logan BR; Griffith LM; Buckley RH; Parrott RE; Dvorak CC; Kapoor N; Hanson IC Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med, 2014.PMID 25075835
- [3]McDonald-McGinn DM; Sullivan KE; Marino B; Philip N; Swillen A; Vorstman JA; Zackai EH; Emanuel BS 22q11.2 deletion syndrome. Nat Rev Dis Primers, 2015.PMID 27189754
- [4]Tangye SG; Al-Herz W; Bousfiha A; Cunningham-Rundles C; Franco JL; Holland SM; Klein C; Morio T The Ever-Increasing Array of Novel Inborn Errors of Immunity: an Interim Update by the IUIS Committee. J Clin Immunol, 2021.PMID 33598806
- [5]Bousfiha A; Jeddane L; Picard C; Ailal F; Bobby Gaspar H; Al-Herz W; Chatila T; Crow YJ The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol, 2018.PMID 29226301
- [6]Poli MC; Aksentijevich I; Bousfiha AA; Cunningham-Rundles C; Hambleton S; Klein C; Morio T; Picard C Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Hum Immun, 2025.PMID 41608114
- [7]Notarangelo LD Genetically-determined defects of T cell development. Allergy Asthma Proc, 2024.PMID 39294907
- [8]Óskarsdóttir S; Boot E; Crowley TB; Loo JCY; Arganbright JM; Armando M Updated clinical practice recommendations for managing children with 22q11.2 deletion syndrome. Genet Med, 2023.PMID 36729053
- [9]Medical Advisory Committee of the Immune Deficiency Foundation; Shearer WT; Fleisher TA; Buckley RH; Ballas Z; Ballow M Recommendations for live viral and bacterial vaccines in immunodeficient patients and their close contacts. J Allergy Clin Immunol, 2014.PMID 24582311
- [10]Lankester AC; Neven B; Mahlaoui N; von Asmuth EGJ; Courteille V; Alligon M Hematopoietic cell transplantation in severe combined immunodeficiency: The SCETIDE 2006-2014 European cohort. J Allergy Clin Immunol, 2022.PMID 34718043
- [11]Kreins AY; Dhalla F; Flinn AM; Howley E; Ekwall O; Villa A European Society for Immunodeficiencies guidelines for the management of patients with congenital athymia. J Allergy Clin Immunol, 2024.PMID 39303894
- [12]van Oers NSC; Sullivan KE The systemic effects of 22q11.2 deletion syndrome on immunity. J Hum Immun, 2026.PMID 41608124