Paeds · cardiology
Aortic and pulmonary stenosis
Also known as congenital aortic stenosis · congenital pulmonary stenosis · valvar aortic stenosis · valvar pulmonary stenosis · bicuspid aortic valve · critical aortic stenosis · critical pulmonary stenosis
A fellowship approach to congenital aortic and pulmonary stenosis: classifying the obstruction by anatomic level (valvar, subvalvar, supravalvar), grading severity by echo peak gradient, recognising the duct-dependent critical neonate, and matching the intervention to the lesion — balloon valvuloplasty as first-line for valvar disease, the Ross procedure for the aortic valve that needs replacement, and syndromic associations from bicuspid aortic valve through Noonan and Williams syndromes.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A murmur heard on the routine newborn or six-week check, a systolic ejection click at the upper left sternal border in an asymptomatic toddler, or a grey, collapsed neonate with a duct that has just closed — these are the three faces of congenital outflow-tract obstruction that a general paediatrician will meet. The fellowship task is to classify the level, grade the severity, recognise the duct-dependent infant, and time the intervention so that the child is neither exposed to unnecessary procedures nor lost to preventable complications. [3] [1]
The three levels — Valve, Subvalve, Supravalve
Overview & Definition
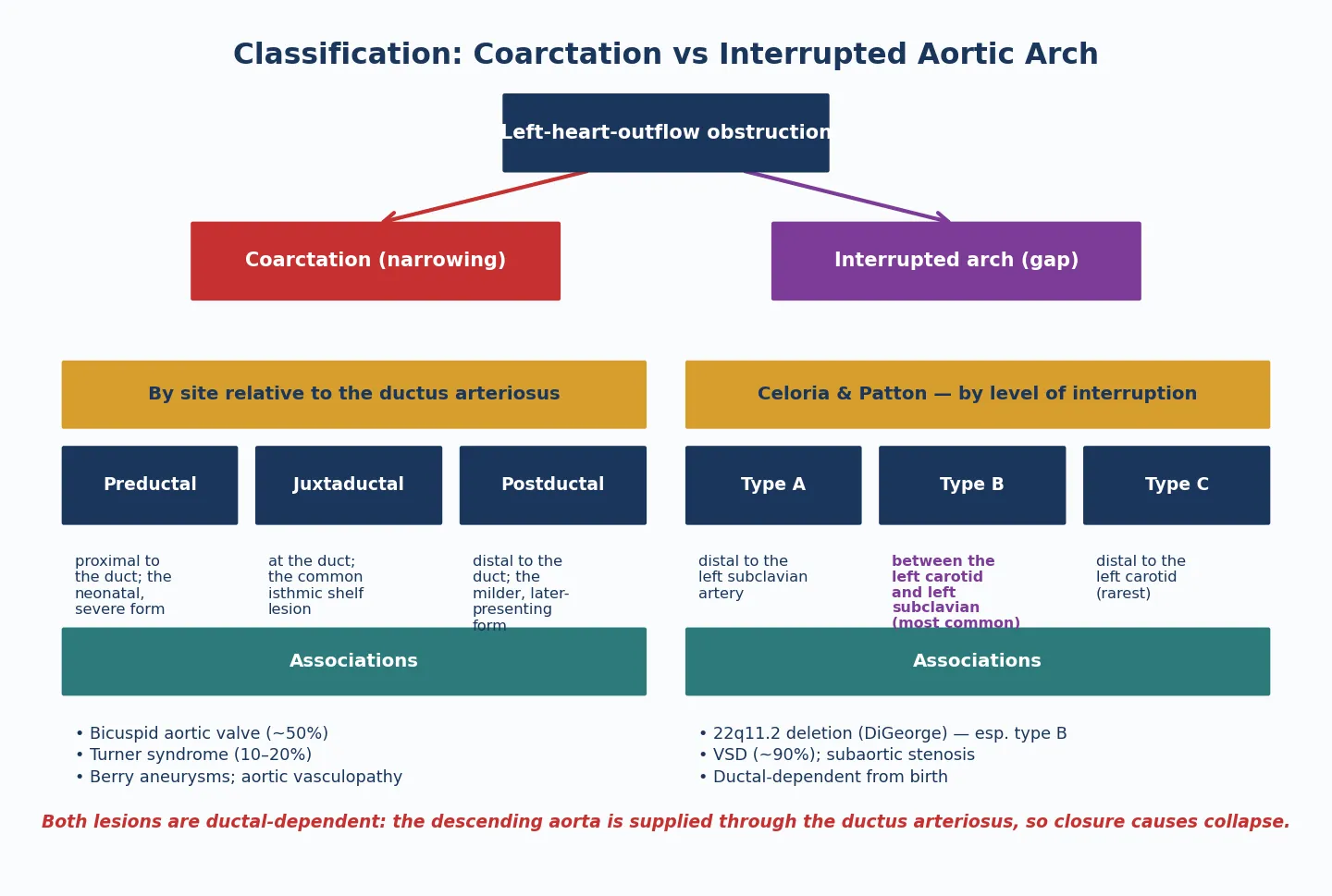
Congenital aortic stenosis and congenital pulmonary stenosis are fixed anatomical narrowings of the left and right ventricular outflow tracts respectively, present from birth and graded by the pressure gradient they impose on ventricular ejection. They are among the commonest forms of congenital heart disease, together accounting for a substantial fraction of all structural cardiac lesions, and they span a spectrum from a silently mild murmur detected at a routine check to a duct-dependent critical obstruction that kills the neonate within hours of ductal closure. [3] [1]
The definition that earns marks is anatomical, not haemodynamic. The lesion is named for the level at which it sits — valvar, subvalvar, or supravalvar — and the severity is a separate, graded axis measured by echocardiographic peak systolic gradient. This dual classification matters because the level determines the intervention and the syndrome, while the severity determines the urgency. A bicuspid aortic valve with a gradient of 25 mmHg is observed; the same valve at 80 mmHg with syncope is an interventional emergency. [1] [4]
The clinical importance of the topic rests on three pillars. First, these are common lesions that every paediatrician will encounter as a murmur, and the ability to distinguish a flow murmur from a pathological ejection systolic murmur with a click is a core skill. Second, the critical neonatal forms are time-sensitive emergencies that demand prostaglandin therapy before the duct closes. Third, the long-term trajectory — restenosis after intervention, aortic root dilation in bicuspid aortic valve, right-ventricular failure in untreated pulmonary stenosis, and the lifelong need for cardiology follow-up — is what makes these conditions chronic, not acute, problems. [2] [3]
Classification
The classification of outflow-tract stenosis follows the blood: from the ventricle outward, the obstruction can be subvalvar, valvar, or supravalvar. Each level carries a characteristic morphology, a characteristic syndrome, and a characteristic intervention, and a fellowship answer earns depth by connecting the three. [1] [4]
Aortic stenosis at the valvar level is the commonest form and is usually a bicuspid aortic valve. The valve has two leaflets instead of three, the leaflets are thickened and dome-shaped in systole, and the obstruction is progressive because the abnormal leaflets calcify and stiffen over decades. In neonates with critical aortic stenosis the valve is often unicuspid or severely dysplastic, producing profound obstruction from birth. Subvalvar aortic stenosis is typically a discrete fibromuscular membrane or a tunnel-like narrowing of the left ventricular outflow tract, and it is associated with Shone complex when it coexists with mitral and aortic coarctation lesions. Supravalvar aortic stenosis is a narrowing above the sino-tubular junction and is the hallmark cardiac lesion of Williams syndrome, caused by elastin (ELN) deletion on chromosome 7. [4] [9]
Pulmonary stenosis follows the same logic. Valvar pulmonary stenosis is the commonest form, with fused or thickened commissures producing a domed valve that narrows the orifice. A distinctive subtype is the dysplastic pulmonary valve — thickened, immobile, myxomatous leaflets without commissural fusion — which is characteristic of Noonan syndrome and responds poorly to balloon valvuloplasty. Supravalvar and branch pulmonary stenosis narrows the main pulmonary artery or its branches and is syndromic: Williams syndrome, Noonan syndrome, Alagille syndrome, and congenital rubella all produce peripheral pulmonary stenosis. Subvalvar or infundibular pulmonary stenosis is usually secondary, most often as part of tetralogy of Fallot. [8] [12]
Epidemiology & Risk Factors
Congenital aortic and pulmonary stenosis are among the commonest congenital cardiac lesions. Isolated pulmonary stenosis accounts for roughly 7 to 10 per cent of all congenital heart disease, and bicuspid aortic valve alone — the leading cause of valvar aortic stenosis — affects approximately 1 to 2 per cent of the population, making it the single commonest congenital cardiac malformation. The combined burden of outflow-tract obstruction represents a large fraction of all structural heart disease seen in paediatric practice. [3] [4]
The strongest risk factor is genetic. Bicuspid aortic valve is familial in a substantial proportion of cases, with autosomal dominant inheritance and incomplete penetrance, which is why first-degree relatives of a child with a bicuspid valve should be screened with echocardiography. Supravalvar aortic stenosis is caused by elastin haploinsufficiency in Williams syndrome, a microdeletion at 7q11.23, and Noonan syndrome — a RASopathy caused by PTPN11 and related mutations — produces both valvar pulmonary stenosis and hypertrophic cardiomyopathy. Alagille syndrome, from JAG1 or NOTCH2 mutations, causes peripheral pulmonary stenosis alongside bile-duct paucity and the characteristic facies. [8] [9]
Maternal rubella infection remains a recognised cause of peripheral pulmonary stenosis and patent ductus arteriosus in regions without universal vaccination, and it is a reminder that congenital outflow obstruction can be acquired in utero as well as inherited. The risk factors that shape severity and presentation include the degree of obstruction at birth — which determines whether the neonate is critical and duct-dependent — and the rate of progression, which is faster in subvalvar aortic stenosis than in valvar disease. [1] [3]
Pathophysiology
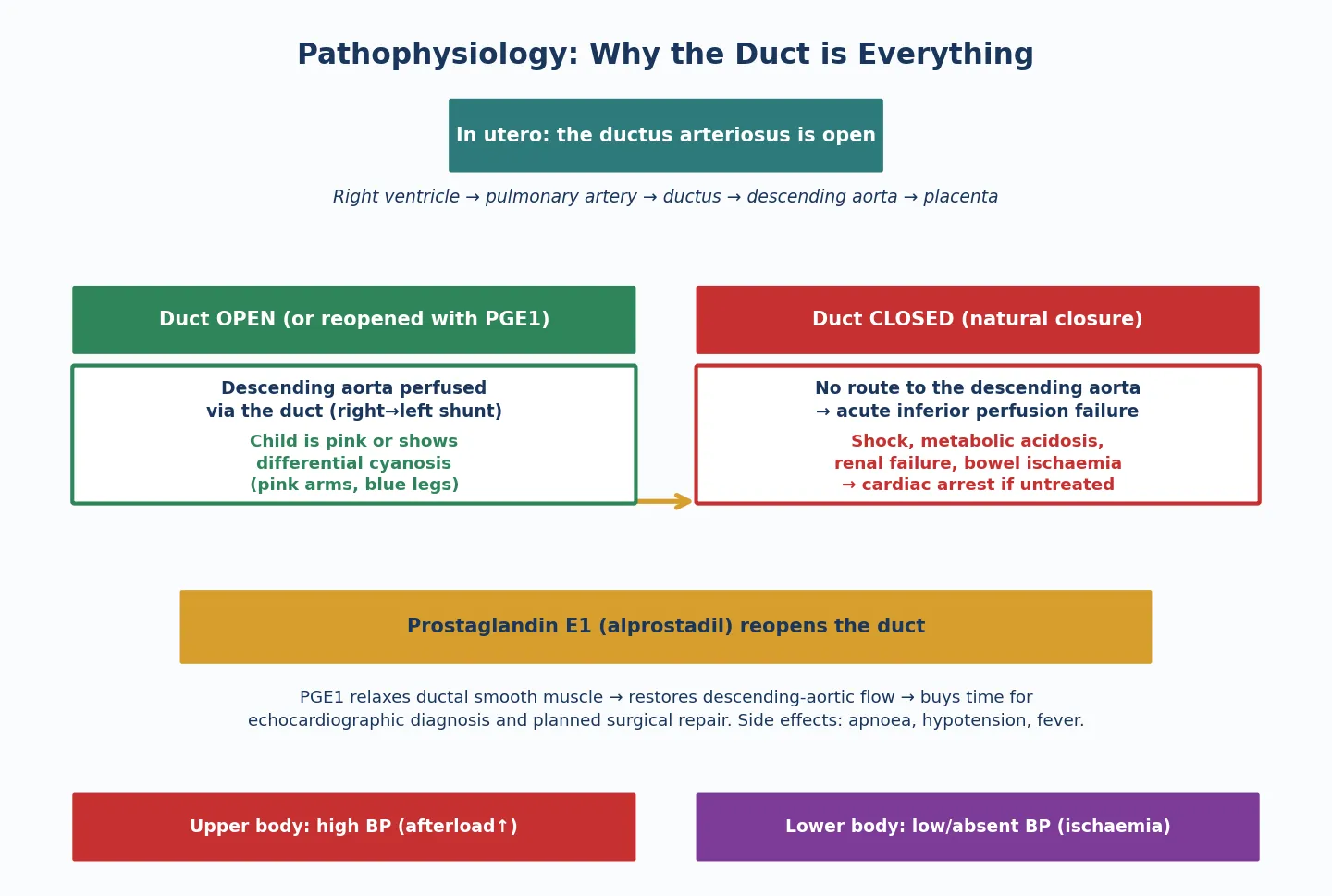
The pathophysiology of outflow-tract stenosis is the pathophysiology of pressure overload, and it differs between the left and right heart in ways that matter clinically. A fixed narrowing at any level raises the pressure the ventricle must generate to eject blood, and the ventricle responds with concentric hypertrophy — increased wall thickness without chamber dilation — that preserves systolic function but reduces compliance and raises filling pressures. [1] [6]
In aortic stenosis the left ventricle hypertrophies, generating pressures that may exceed 200 mmHg in severe disease, and the thickened muscle has increased oxygen demand while the raised systolic pressure compresses the subendocardial coronary circulation. The result is subendocardial ischaemia, which is the mechanism behind exertional chest pain and syncope — the symptoms that mark a child at risk of sudden cardiac death. In critical neonatal aortic stenosis the left ventricle cannot generate adequate cardiac output, and systemic perfusion becomes dependent on the ductus arteriosus delivering right-ventricular blood to the descending aorta. When the duct closes, the neonate collapses into cardiogenic shock with metabolic acidosis. [6] [1]
In pulmonary stenosis the right ventricle hypertrophies in response to the raised afterload, and right-ventricular systolic function is preserved for years because the hypertrophy is an appropriate adaptation. The problems arise when the right-ventricular pressure approaches or exceeds systemic pressure: the hypertrophied muscle becomes non-compliant, right-atrial pressure rises, and if a patent foramen ovale or atrial septal defect is present, the right-to-left shunt produces cyanosis. In critical pulmonary stenosis the right ventricle is severely hypertrophied with a small cavity, pulmonary blood flow is duct-dependent, and ductal closure produces profound cyanosis without shock. [12] [11]
The severity grading — mild, moderate, severe — is calibrated to these haemodynamic consequences and is measured by the peak instantaneous systolic gradient on continuous-wave Doppler echocardiography. The commonly used thresholds are mild below 40 mmHg, moderate 40 to 64 mmHg, and severe above 64 mmHg, with the mean gradient also reported. The gradient is not a perfect measure of severity because it depends on ventricular function — a failing ventricle generates a lower gradient across a severe obstruction — but in a child with preserved function it is the single most useful number in the topic. [1] [2]
Clinical Presentation
The clinical presentation spans a spectrum from the asymptomatic child with an incidental murmur to the critically ill neonate in shock, and the fellowship answer frames it by severity and age. The commonest presentation is a well child with a systolic murmur heard at a routine newborn examination, six-week check, or school entry screening, and the task is to distinguish a pathological outflow-tract murmur from a benign innocent murmur. [3] [1]
The classic auscultatory findings of valvar aortic stenosis are a systolic ejection murmur at the upper right sternal border radiating to the carotids and suprasternal notch, preceded by an ejection click that is heard best at the apex and does not vary with respiration. The click is the stenotic valve opening, and its presence localises the lesion to the valve. A thrill is palpable at the upper right sternal border or suprasternal notch in moderate to severe disease. Valvar pulmonary stenosis produces a systolic ejection murmur at the upper left sternal border radiating to the back and left infraclavicular area, with an ejection click that varies with respiration — heard best in expiration because the pulmonary valve opens closest to the chest wall then. The wider the split of the second heart sound, the more severe the pulmonary stenosis, because delayed pulmonary valve closure reflects prolonged right-ventricular ejection. [1] [2]
Symptoms appear in moderate to severe disease and are the warning signs. Exertional chest pain, exertional dyspnoea, and syncope in aortic stenosis signal subendocardial ischaemia and identify the child at risk of sudden death. In pulmonary stenosis, exertional dyspnoea and right-heart failure symptoms appear late, and cyanosis from right-to-left atrial shunting is the sign that the right-ventricular pressure has exceeded left-atrial pressure. [6] [11]
The critical neonate is the presentation that must not be missed. A neonate with critical aortic stenosis presents with poor perfusion, grey pallor, weak pulses, tachypnoea, and a metabolic acidosis as the duct closes in the first days of life — the picture of cardiogenic shock that is indistinguishable from sepsis without a high index of suspicion. A neonate with critical pulmonary stenosis presents with profound cyanosis, a loud single second heart sound, and a right-ventricular impulse, because pulmonary blood flow is duct-dependent and the closing duct removes the only source of pulmonary perfusion. Both presentations demand prostaglandin E1, echocardiography, and urgent cardiology referral. [6] [12]
Differential Diagnosis
The differential diagnosis of a systolic ejection murmur at the upper sternal border is broad, and the fellowship task is to distinguish outflow-tract stenosis from the benign and the mimics. The innocent murmurs — pulmonary flow murmur, Still's murmur, and peripheral pulmonary stenosis of the newborn — are the commonest confounders, and they are distinguished by their soft quality, absence of a click, normal second heart sound, and the absence of a gradient on echocardiography. [3] [1]
Within pathological murmurs, the anatomic level of the lesion is the key discriminator. An aortic stenosis murmur at the upper right sternal border with a click at the apex is distinguished from a pulmonary stenosis murmur at the upper left sternal border with a respiratory-varying click and a widely split second sound. Subvalvar aortic stenosis produces a murmur without a click, because the obstruction is below the valve and the valve itself opens normally. Supravalvar aortic stenosis produces a murmur that is louder in the right suprasternal area — the so-called right-sided dominance — because the jet is directed into the right innominate artery. [4] [9]
The syndromic context narrows the differential powerfully. A child with elfin facies, a sociable personality, developmental delay, and hypercalcaemia has Williams syndrome with supravalvar aortic stenosis and peripheral pulmonary stenosis. A child with short stature, webbed neck, ptosis, and a chest deformity has Noonan syndrome with valvar pulmonary stenosis from a dysplastic valve and possible hypertrophic cardiomyopathy. A child with a heart murmur, biliary atresia or cholestasis, vertebral anomalies, and the characteristic facies has Alagille syndrome with peripheral pulmonary stenosis. Recognising the syndrome predicts the lesion and the management. [8] [9]
The differential of the collapsed neonate with shock or cyanosis includes sepsis, metabolic disease, and other duct-dependent lesions — hypoplastic left heart syndrome, coarctation of the aorta, and transposition of the great arteries. The four-extremity blood pressure, pre- and post-ductal saturations, and the response to prostaglandin E1 are the discriminators, and echocardiography is the definitive investigation. A fellowship answer does not wait for the differential to close before starting prostaglandin if a duct-dependent lesion is suspected. [6] [2]
| Feature | Valvar AS | Valvar PS | Innocent flow | Subvalvar AS |
|---|---|---|---|---|
| Location | Upper RSB | Upper LSB | Upper LSB / diffuse | Upper RSB |
| Ejection click | Yes (apex, fixed) | Yes (LSB, varies) | No | No |
| S2 splitting | Normal | Wide (delayed P2) | Normal | Normal |
| Radiates to | Carotids | Back / infraclavicular | Nowhere specific | Carotids |
| Echo gradient | Raised | Raised | None | Raised |
Clinical & Bedside Assessment
The recognition move runs off the murmur and the clinical context, and the bedside task is to grade the severity and recognise the critical neonate. The history gathers the discriminators: the presence and timing of the murmur, the child's symptoms on exertion — chest pain, dyspnoea, syncope — the family history of congenital heart disease or sudden death, and the syndromic features that point to Williams, Noonan, or Alagille syndrome. [1] [3]
Examination is systematic and focused. Palpate the precordium for a thrill at the upper right or left sternal border, a sign of at least moderate obstruction. Auscultate for the location and radiation of the ejection systolic murmur, the presence and characteristics of the ejection click, and the splitting of the second heart sound. Check the pulses in all four limbs — weak or absent femoral pulses suggest coarctation, and differential blood pressures between the arms and legs narrow the lesion. Measure pre- and post-ductal oxygen saturations in any neonate with suspected obstruction. Look for the syndromic facies and features that point to the underlying diagnosis. [2] [9]
The critical neonate is assessed differently and with urgency. A neonate with poor perfusion, weak pulses, tachypnoea, hepatomegaly, and a metabolic acidosis has cardiogenic shock from a duct-dependent left-heart obstruction until proven otherwise. A neonate with profound cyanosis that does not improve with oxygen — the hyperoxia test — has a right-to-left shunt or duct-dependent pulmonary flow. In both, the response is immediate: start prostaglandin E1, secure the airway if apnoea occurs, obtain venous access, check gas and lactate, and arrange urgent echocardiography. [6] [12]
The first paediatric cardiology visit after a murmur is detected gathers the echocardiographic data that drives every subsequent decision. The echocardiogram defines the anatomic level of the obstruction, the valve morphology, the peak and mean gradient, the ventricular function and hypertrophy, the aortic root diameter in bicuspid aortic valve, and any associated lesions. The gradient is the number that drives the management pathway, and a fellowship answer can state it and act on it. [1] [4]
Investigations
Echocardiography is the single definitive investigation, and it answers every clinically relevant question about outflow-tract stenosis. Two-dimensional imaging defines the anatomic level — valvar, subvalvar, or supravalvar — and the valve morphology, including the number of leaflets in aortic stenosis and the presence of dysplasia in pulmonary stenosis. Continuous-wave Doppler measures the peak instantaneous systolic gradient, which is the severity-grading metric, and colour-flow mapping identifies associated regurgitation, shunts, or collateral vessels. [1] [4]
The severity grading by peak gradient is the operational framework. Mild stenosis is a peak gradient below 40 mmHg, moderate is 40 to 64 mmHg, and severe is above 64 mmHg. The mean gradient is also reported and is increasingly used in guidelines, but the peak gradient remains the most widely used threshold in paediatric practice. In aortic stenosis the echocardiogram also measures the aortic root and ascending aorta diameter, because bicuspid aortic valve is associated with aortopathy and progressive root dilation independent of the gradient. [4] [2]
The electrocardiogram is a useful adjunct and shows left or right ventricular hypertrophy with strain patterns in moderate to severe disease, but it is not sensitive enough to exclude significant obstruction. Chest radiography is usually normal in mild disease and may show cardiomegaly, pulmonary oligoaemia in pulmonary stenosis, or pulmonary venous congestion in critical aortic stenosis. Exercise testing is valuable in older children with moderate aortic stenosis to assess functional capacity, blood pressure response, and ST-segment changes — an abnormal exercise test is an indication for intervention even if the resting gradient is borderline. [1] [2]
Cardiac catheterisation is primarily therapeutic rather than diagnostic in this topic. The peak-to-peak gradient measured at catheterisation confirms the severity before balloon valvuloplasty, and angiography defines the anatomy for the intervention. Cardiac magnetic resonance imaging is reserved for complex anatomy, particularly branch pulmonary stenosis, and for assessing right-ventricular volume and function in chronic pulmonary stenosis. Genetic testing is indicated when a syndromic cause is suspected — FISH or chromosomal microarray for Williams syndrome, RASopathy panels for Noonan syndrome — because the genetic diagnosis informs the management and the family counselling. [8] [9]
Management — Resuscitation
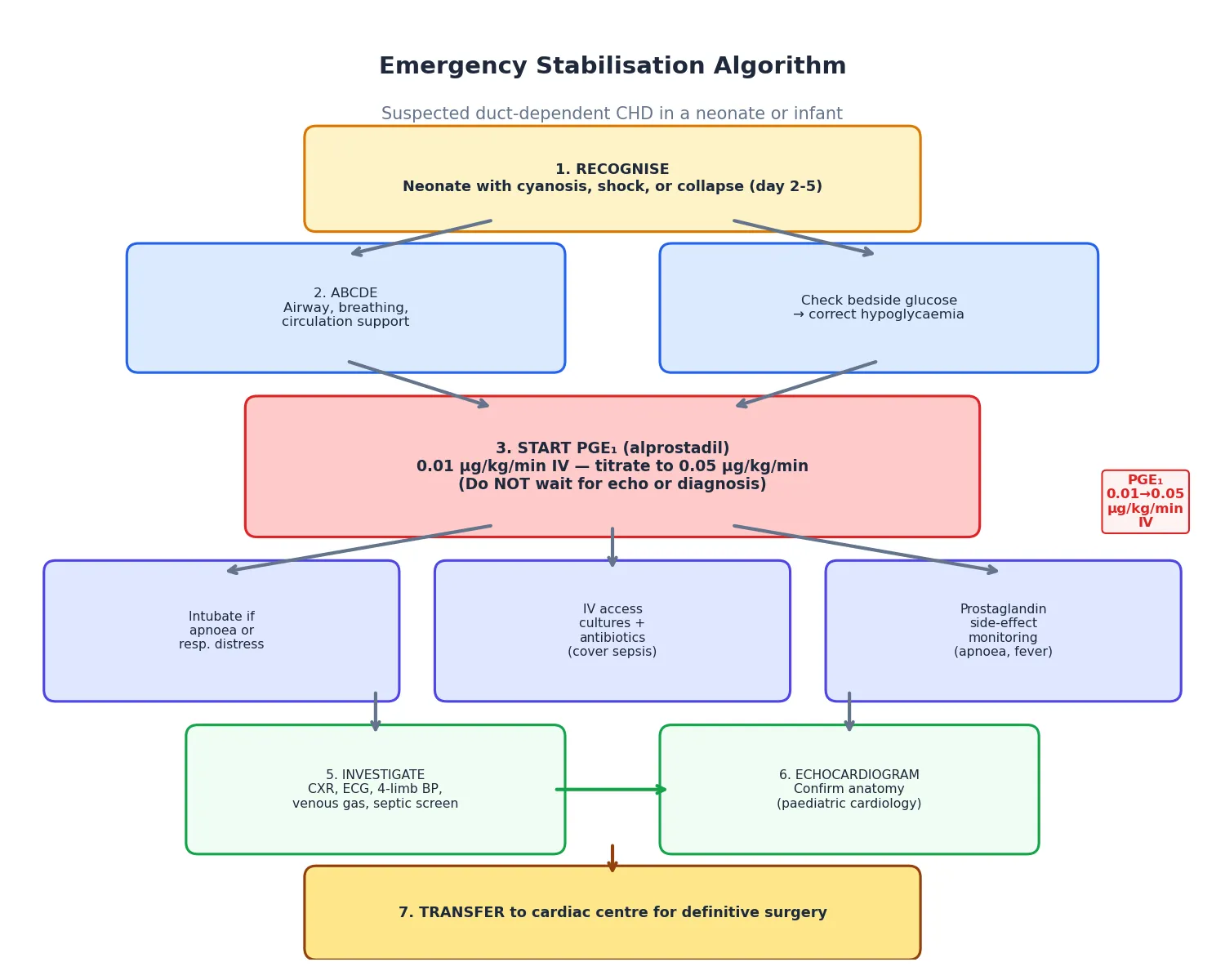
Resuscitation in outflow-tract stenosis applies to the critical neonate, and the intervention is prostaglandin E1. In critical aortic stenosis the ductus delivers right-ventricular blood to the descending aorta, maintaining systemic perfusion that the obstructed left ventricle cannot provide. In critical pulmonary stenosis the ductus delivers blood to the pulmonary circulation, maintaining oxygenation that the obstructed right ventricle cannot support. When the duct closes, both neonates deteriorate catastrophically, and prostaglandin E1 at 0.01 to 0.05 mcg per kg per minute is the bridge to definitive intervention. [6] [12]
The resuscitation of the critical neonate is a sequence. Recognise the duct-dependent physiology from the clinical picture — shock or cyanosis in the first days of life, poor perfusion, abnormal pulses, hepatomegaly, metabolic acidosis. Start prostaglandin E1 immediately, without waiting for echocardiographic confirmation, because the cost of unnecessary prostaglandin is far lower than the cost of a missed duct-dependent lesion. Secure the airway, because prostaglandin-induced apnoea is common and many neonates require intubation. Obtain venous access, check blood gas and lactate, start inotropes if myocardial dysfunction is present, and arrange urgent echocardiography and cardiology referral. [6] [2]
The definitive resuscitation intervention is balloon valvuloplasty for the critical neonate with valvar obstruction, performed urgently once the child is stabilised on prostaglandin. In critical aortic stenosis the balloon dilates the stenotic valve to relieve the gradient and allow the left ventricle to support the systemic circulation; the alternative is a hybrid or surgical approach if the left ventricle is too small or hypoplastic. In critical pulmonary stenosis the balloon opens the stenotic valve, and if the right ventricle is too small or hypoplastic, a systemic-to-pulmonary shunt may be needed until the right ventricle grows. [5] [12]
The older child with symptomatic severe obstruction — chest pain, syncope, or an abnormal exercise test — is a different resuscitation scenario. The intervention is elective or urgent balloon valvuloplasty or surgery depending on the lesion, and the goal is to relieve the obstruction before a sudden-death event. The child should be advised to avoid competitive sport and strenuous exertion until the obstruction is relieved. [1] [6]
Management — Definitive & Stepwise
Definitive management is gradient-guided and matched to the anatomic level of the obstruction. Mild disease (peak gradient below 40 mmHg) is managed with clinical surveillance and serial echocardiography every one to three years for aortic stenosis and every two to five years for pulmonary stenosis, with no intervention unless the gradient progresses. Moderate and severe disease are referred to the interventional cardiologist. [1] [2]
For valvar aortic stenosis, balloon aortic valvuloplasty is the first-line intervention in children and adolescents. The balloon is advanced across the stenotic valve and inflated to split the fused commissures, reducing the gradient. The Valvuloplasty and Angioplasty of Congenital Anomalies Registry data established the predictors of immediate success and the complication profile, and balloon valvuloplasty carries a low rate of major complications, though aortic regurgitation is a recognised consequence. The alternative for the aortic valve that cannot be ballooned or that needs replacement is the Ross procedure, in which the patient's own pulmonary valve is transferred to the aortic position and a homograft replaces the pulmonary valve. The Ross procedure is preferred in growing children because the autograft grows with the child and avoids the need for anticoagulation, though long-term follow-up shows a rate of autograft dilation and homograft stenosis that requires reoperation. [5] [7]
For valvar pulmonary stenosis, balloon pulmonary valvuloplasty is the first-line and definitive intervention, and it is one of the most successful procedures in congenital interventional cardiology. The long-term outcome data show sustained gradient reduction with low rates of restenosis and a low complication rate, and the dysplastic Noonan valve is the main exception — it responds poorly to ballooning and often needs surgical valvotomy. Subvalvar and supravalvar obstruction are surgical: subvalvar aortic stenosis is resected, supravalvar aortic stenosis is patched or reconstructed, and branch pulmonary stenosis is stented or surgically augmented. [10] [8]
Long-term surveillance is universal after any intervention. Restenosis is common after balloon valvuloplasty and may require repeat intervention. Aortic regurgitation is the late concern after aortic valvuloplasty and after the Ross procedure, and progressive aortic root dilation in bicuspid aortic valve requires monitoring regardless of the valve gradient. The child with a prosthetic valve or prosthetic material within six months of a procedure needs endocarditis prophylaxis before dental and selected procedures, and all children with significant obstruction or prosthetic valves need lifelong cardiology follow-up with structured transition to adult congenital heart disease services. [1] [2]
Specific Subtypes & Scenarios
The bicuspid aortic valve is the single most important subtype because it is the commonest congenital cardiac lesion and the commonest cause of valvar aortic stenosis. The valve has two leaflets instead of three — usually a right and left coronary leaflet fused — and it may be silent in childhood or produce a murmur and click that brings the child to attention. The valve may stenose, regurgitate, or both, and it is associated with aortopathy — coarctation of the aorta in up to 10 per cent of cases and progressive aortic root and ascending aorta dilation that is independent of the valve gradient. First-degree relatives should be screened because of the familial pattern, and the child needs lifelong surveillance even if the valve gradient is mild. [4] [1]
Williams syndrome — the 7q11.23 microdeletion causing elastin haploinsufficiency — produces supravalvar aortic stenosis, peripheral pulmonary artery stenosis, and a characteristic phenotype of elfin facies, stellate irises, a loquacious and overly friendly personality, developmental delay, infantile hypercalcaemia, and a hoarse voice. The cardiac lesions are the cause of mortality, and the supravalvar aortic stenosis may be progressive. Coronary artery abnormalities — ostial stenosis or coronary dilation — are a recognised feature and a source of sudden death, particularly under anaesthesia or sedation, which makes anaesthetic management high-risk. A fellowship answer names the coronary risk and the need for careful anaesthetic planning. [9] [1]
Noonan syndrome — a RASopathy most commonly from PTPN11 mutations — produces valvar pulmonary stenosis from a dysplastic, thickened, immobile valve that is morphologically distinct from the classic domed commissural-fusion valve. The dysplastic valve responds poorly to balloon valvuloplasty and often needs surgical valvotomy, and it may coexist with hypertrophic cardiomyopathy, which is the other cardiac feature of the syndrome. The phenotype includes short stature, webbed neck, ptosis, low-set ears, a broad or shield chest, and developmental delay. Recognising the syndrome predicts the valve morphology and the response to intervention. [8] [12]
The critical neonate is the scenario where recognition and timing determine outcome. Critical aortic stenosis presents with shock as the duct closes, and the left ventricle may be too small to support the systemic circulation after relief of the obstruction — in which case a single-ventricle (Norwood) pathway may be needed. Critical pulmonary stenosis presents with cyanosis, and the right ventricle may be too small to accept the full venous return — in which case a systemic-to-pulmonary shunt is needed until the right ventricle grows. The distinction between a biventricular repair and a single-ventricle pathway is made by the echocardiographic assessment of ventricular size, and it is the decision that defines the child's entire surgical future. [6] [12]
Why the Ross procedure is preferred in children — and what its limitations are
The Ross procedure replaces the diseased aortic valve with the patient's own pulmonary autograft and places a homograft in the pulmonary position. Its advantage in a growing child is that the autograft is living tissue that grows with the child and does not require anticoagulation, unlike a mechanical valve. Its limitation is that the autograft may dilate over time, producing aortic regurgitation, and the pulmonary homograft stenoses and needs replacement, typically every 10 to 15 years. Long-term data from the Ross versus aortic valve repair comparison show that both are viable options in children, and the choice depends on valve morphology and surgical expertise. Naming the trade-offs is what distinguishes a fellowship answer from a checklist. [7] [1]
Complications & Pitfalls
Untreated severe aortic stenosis carries a risk of sudden cardiac death from ventricular arrhythmia precipitated by subendocardial ischaemia — a child who collapses during exercise with a known high gradient is the scenario every paediatrician fears, and it is what justifies intervention even in the asymptomatic child. Untreated pulmonary stenosis takes a slower course, progressing over years to right-ventricular failure and right-to-left shunting as the hypertrophied right ventricle eventually decompensates. The procedural complications are the price of intervention — aortic and pulmonary regurgitation after valvuloplasty, autograft dilation and homograft stenosis after the Ross procedure — and the long-term trajectory is one of expected reoperation and lifelong surveillance. [1] [6]
Procedural complications are the price of intervention. Balloon aortic valvuloplasty produces aortic regurgitation in a proportion of cases, and severe regurgitation may require valve replacement. Balloon pulmonary valvuloplasty produces pulmonary regurgitation, which is usually well tolerated in childhood but may cause right-ventricular dilation and dysfunction over decades and require pulmonary valve replacement — typically with a percutaneous valve in the adult. The Ross procedure carries the risk of autograft dilation and homograft stenosis requiring reoperation. Surgical resection of subvalvar aortic stenosis carries a risk of complete heart block and of recurrence — the membrane grows back in a subset of children. [5] [7]
The management pitfalls are the errors that cost marks and harm children. Failing to recognise the critical neonate and treating the presentation as sepsis without starting prostaglandin is the cardinal error — the neonate deteriorates and may die before the duct-dependent physiology is identified. Delaying intervention in a child with symptomatic severe aortic stenosis exposes the child to the sudden-death risk. Performing balloon valvuloplasty on a dysplastic Noonan valve when surgical valvotomy is the correct intervention wastes a procedure and exposes the child to unnecessary radiation. Forgetting to screen first-degree relatives of a child with a bicuspid aortic valve misses affected family members. And losing the child to cardiology follow-up after a successful intervention allows restenosis, regurgitation, or aortic dilation to progress silently. [4] [2]
Prognosis & Disposition
The prognosis of congenital outflow-tract stenosis is excellent when the lesion is recognised, graded, and managed in a timely way. Mild disease is compatible with a normal life with surveillance, and children with mild valvar pulmonary stenosis have a natural history of stability or even regression of the gradient over time. Moderate and severe disease that is intervened upon appropriately has a good long-term outlook, with most children achieving normal or near-normal exercise tolerance and quality of life. [11] [1]
The qualifications to that optimism are what make the prognosis honest. Severe aortic stenosis carries a residual sudden-death risk even after successful intervention, and all children with significant obstruction need lifelong cardiology follow-up. The Ross procedure and balloon valvuloplasty are not cures — they are interventions that buy time, and reoperation is expected over the lifespan. Bicuspid aortic valve is a lifelong condition that requires surveillance for stenosis, regurgitation, and aortopathy even when the initial presentation is mild. The child with a prosthetic valve needs anticoagulation management and endocarditis prophylaxis. [4] [7]
The general paediatrician owns the recognition and the coordination, and the disposition is shared, structured care. Paediatric cardiology drives the diagnostic echocardiography, the severity grading, the intervention, and the long-term surveillance. The interventional cardiologist performs the balloon valvuloplasty, and the congenital cardiac surgeon performs the Ross procedure, the surgical valvotomy, and the resections. The critical neonate is managed in a neonatal or cardiac intensive care unit with prostaglandin support. Transition to adult congenital heart disease services is a structured, planned handover, not a discharge, and it should begin in early adolescence. [1] [2]
Recurrence-risk counselling applies to the familial and syndromic forms. Bicuspid aortic valve is autosomal dominant with incomplete penetrance, and first-degree relatives have a 9 to 10 per cent chance of being affected. Williams and Noonan syndromes are autosomal dominant with variable expressivity, and the recurrence risk depends on whether a parent is affected. Non-syndromic valvar pulmonary stenosis is usually sporadic with a low recurrence risk. The counselling addresses not only the next pregnancy but the family's understanding of the child's lifelong trajectory. [4] [8]
Special Populations
Congenital outflow-tract stenosis interacts with the child's social, cultural, and geographic context in ways that shape detection, access, and long-term outcomes. A child in a rural or remote community is further from paediatric cardiology services, and the murmur detected at a remote clinic may face delays in echocardiographic confirmation and intervention. Telehealth cardiology and retrieval services extend specialist access, and a low threshold to refer any neonate with a murmur and poor perfusion for assessment is the safeguard against missed critical disease. [1] [2]
Indigenous children in Australia and Aotearoa New Zealand face a higher prevalence of rheumatic heart disease, which can coexist with or mimic congenital outflow-tract obstruction, and reduced access to specialist services in remote communities. The consequences of delayed detection and intervention fall hardest where access is poorest, and outreach echocardiography and cardiology clinics in Indigenous health services are part of the solution. Migrant and refugee families may arrive with an uncertain cardiac history, an unreported murmur, or an incomplete intervention record, and the task is to reconstruct the history, confirm the anatomy by echocardiography, and integrate the child into surveillance. [3] [8]
Socioeconomic disadvantage shapes the practical feasibility of lifelong surveillance. The child with a Ross procedure needs serial echocardiography and, eventually, reoperation; the family needs transport, appointment coordination, and the health literacy to understand why a child who looks well needs ongoing cardiology care. Structuring the surveillance around a single coordinated visit, linking the family to support services, and using telehealth to reduce travel all improve engagement. The child with a syndromic form — Williams, Noonan, Alagille — is a population in their own right, because the syndrome adds developmental, educational, and behavioural needs that must be addressed alongside the cardiac management. [9] [8]
The adolescent with congenital outflow-tract stenosis faces the twin challenges of adherence and transition. A teenager who feels well may not understand why they need continued cardiology follow-up, and the risk of loss to follow-up is highest in the transition years. Structured transition — beginning in early adolescence, with a named adult cardiologist, a documented summary, and the young person's active participation in the handover — is the intervention that prevents the young adult from re-presenting with a preventable complication. [1] [2]
Evidence, Guidelines & Regional Differences
The evidence base rests on two major guideline documents and a body of registry and cohort data. The 2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease is the most comprehensive operational reference for the adolescent and adult with outflow-tract stenosis, covering the severity grading, the intervention thresholds, the surveillance intervals, the endocarditis prophylaxis indications, and the sport participation guidance. The 2020 ESC Guidelines for the management of adult congenital heart disease provide the European parallel, and the two documents are broadly concordant on the management of valvar aortic and pulmonary stenosis. [1] [2]
The interventional evidence is anchored by the Valvuloplasty and Angioplasty of Congenital Anomalies Registry, which established the predictors of success and the complication profile of balloon aortic valvuloplasty in children. The long-term outcomes of balloon pulmonary valvuloplasty — sustained gradient reduction, low restenosis, and low complication rates — are well documented in cohort studies. The Ross procedure has been compared with aortic valve repair in children, and both are viable options with different trade-offs, and the choice depends on valve morphology and surgical expertise. [5] [10] [7]
The epidemiological evidence is anchored by the Hoffman and Kaplan 2002 study of the incidence of congenital heart disease, which remains the reference for the prevalence of pulmonary stenosis and bicuspid aortic valve. The natural history of isolated pulmonary valve stenosis — stability or regression of mild gradients, progression of moderate gradients — is documented in Doppler echocardiographic cohort studies that inform the surveillance strategy. The critical neonatal literature — preoperative management of critical aortic valvar stenosis, the approach to critical pulmonary stenosis — frames the emergency pathway. [3] [11] [6] [12]
Where the evidence is weak or evolving, a fellowship answer says so. The optimal timing of intervention in moderate asymptomatic aortic stenosis, the threshold for aortic root surgery in bicuspid aortic valve, the long-term durability of the Ross autograft, and the role of percutaneous pulmonary valve replacement in the adult are areas of evolving practice. The management of the dysplastic Noonan valve and the choice between biventricular and single-ventricle pathways in the critical neonate are decisions made by the multidisciplinary team on individual anatomy, not by guideline algorithm alone. [8] [6]
In Australia and Aotearoa New Zealand, congenital outflow-tract stenosis is managed within the paediatric cardiology network centred on the major children's hospitals, with shared care involving general paediatricians and primary care across rural and remote regions. Balloon valvuloplasty is the first-line intervention for valvar disease and is performed in all major paediatric cardiac centres. The Ross procedure is available for the aortic valve needing replacement, and surgical expertise in congenital cardiac surgery is concentrated in the tertiary centres. Telehealth extends specialist consultation to remote communities, and retrieval services transport the critical neonate to the cardiac centre on prostaglandin support. Endocarditis prophylaxis follows the international guidelines, and sport participation is guided by the eligibility recommendations. [1] [2]
Exam Pearls
A fellowship candidate answering on aortic and pulmonary stenosis should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for; the traps are where easy marks are lost. [1] [3]
Anchor one: classify by level. Every stenotic lesion is valvar, subvalvar, or supravalvar. The level predicts the syndrome (bicuspid valve, Williams, Noonan), the echo view, and the intervention (balloon for valvar, surgery for subvalvar and supravalvar). [4]
Anchor two: grade by gradient. Mild below 40 mmHg, moderate 40 to 64 mmHg, severe above 64 mmHg. The gradient is the number that drives the management pathway, and it is measured by continuous-wave Doppler echocardiography. [1]
Anchor three: recognise the critical neonate. Critical aortic stenosis is duct-dependent for systemic flow; critical pulmonary stenosis is duct-dependent for pulmonary flow. Both need prostaglandin E1 at 0.01 to 0.05 mcg per kg per minute before the duct closes. [6] [12]
Anchor four: match the intervention. Balloon valvuloplasty is first-line for valvar aortic and pulmonary stenosis. Surgery for subvalvar and supravalvar obstruction, dysplastic Noonan valves, and residual gradients. The Ross procedure for the aortic valve needing replacement in a growing child. [5] [7]
Anchor five: surveil for life. Restenosis, regurgitation, aortic root dilation, and reoperation are expected, not exceptions. Lifelong cardiology follow-up with structured transition to adult congenital heart disease services is the standard. [1] [2]
The three traps to avoid are treating the collapsed neonate as sepsis without considering duct-dependent obstruction, ballooning a dysplastic Noonan valve when surgical valvotomy is needed, and forgetting that bicuspid aortic valve is familial with associated aortopathy that needs screening. Bicuspid aortic valve affects 1 to 2 per cent of the population, pulmonary stenosis accounts for 7 to 10 per cent of all congenital heart disease, the ejection click localises the lesion to the valve, and prostaglandin E1 is the first drug in the critical neonate — the high-yield facts a candidate holds. [3] [4]
References
- [1]Stout KK, Daniels CJ, Aboulhosn JA, Bozkurt B, Broberg CS, Colman JM, Crumb SR, Dearani JA, Fuller S, Gurvitz M, Khairy P, Landzberg MJ, Saidi A, Valente AM, Van Hare GF. 2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation, 2019.PMID 30586767
- [2]Baumgartner H, De Backer J, Babu-Narayan SV, Budts W, Chessa M, Diller GP, Lung B, Kluin J, Lang IM, Meijboom F, Moons P, Mulder BJM, Popelová J, Price S, Roos-Hesselink J, Ruel M, Van Dijk APJ, Veldtman GR, Hassan Murad M, Trindade PT, Ruschitzka F, Williams BA, ESC Scientific Document Group. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur Heart J, 2021.PMID 32860028
- [3]Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol, 2002.PMID 12084585
- [4]Spaziani G, Girolami F, Arcieri L, Pollazzi S, Pradella S, Polloni I, Santoro C, la Marca G, Calabri GB, Favilli S, Fedi S, Vergine G, Calabro R, Favilli N, Assanta N, Spina M, Tomasi L, Amodeo A, Rapezzi C, Di Cesare E, Gargiulo GD, Tchana B, Iacomino M, Casadonte F, Fiumana A, Franciosi S, Biondi A, Giglio AF, Drago F, Bisceglia M, Favale S, Tomasi M, Iorio FS, Adorisio R, Squarcia U, De Zorzi A, Castaldi B, Dallapiccola B, Mingarelli R. Bicuspid Aortic Valve in Children and Adolescents: A Comprehensive Review. Diagnostics (Basel), 2022.PMID 35885654
- [5]McCrindle BW. Independent predictors of immediate results of percutaneous balloon aortic valvotomy in children. Valvuloplasty and Angioplasty of Congenital Anomalies Registry. Am J Cardiol, 1996.PMID 8607410
- [6]Affolter JT, Ghanayem NS. Preoperative management of the neonate with critical aortic valvar stenosis. Cardiol Young, 2014.PMID 25647388
- [7]Zhu MZL, Konstantinov IE, Wu DM, Li S, Sleeper LA, Pigula FA, Mayer JE, del Nido PJ, Baird CW. Aortic valve repair versus the Ross procedure in children. J Thorac Cardiovasc Surg, 2023.PMID 37169064
- [8]Linglart L, Gelb BD. Congenital heart defects in Noonan syndrome: Diagnosis, management, and treatment. Am J Med Genet C Semin Med Genet, 2020.PMID 32022400
- [9]Twite MD, Stenquist S, Ing RJ. Williams syndrome. Paediatr Anaesth, 2019.PMID 30811742
- [10]Devanagondi R, Peck D, Sagi J, Gradus-Pizlo I, Shively B, Hoyer MH, Rosenthal GL, Armstrong AK. Long-Term Outcomes of Balloon Valvuloplasty for Isolated Pulmonary Valve Stenosis. Pediatr Cardiol, 2017.PMID 27826708
- [11]Rowland DG, Hammill WW, Allen HD. Natural course of isolated pulmonary valve stenosis in infants and children utilizing Doppler echocardiography. Am J Cardiol, 1997.PMID 9036756
- [12]Latson LA. Critical pulmonary stenosis. J Interv Cardiol, 2001.PMID 12053395