Paeds · cardiology
Atrioventricular septal defect
Also known as AVSD · Atrioventricular canal defect · Endocardial cushion defect · Ostium primum atrial septal defect · Complete atrioventricular canal
Fellowship guide to atrioventricular septal defect — the shared common atrioventricular junction at the crux of the heart, the partial–transitional–complete spectrum, the invariable superior QRS axis, the Down syndrome association, and why complete defects are repaired before six months to prevent irreversible pulmonary vascular disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the four chambers of the heart meeting at a single point — the crux — where the atrial septum, the ventricular septum, and the atrioventricular valves all interlock. In an atrioventricular septal defect, that meeting never happens properly. The endocardial cushions that should fuse to close this junction fail, leaving a single, shared atrioventricular junction that straddles both the atrial and the ventricular septum, with malformed valve leaflets that bridge across the gap. This is why the old names — endocardial cushion defect and atrioventricular canal — still surface in exams, and why the morphology is impossible to understand without holding the crux in your mind. [1]
The definition worth memorising is anatomical and unifying. An AVSD is a congenital heart defect characterised by a deficiency of the atrioventricular septum, a common atrioventricular junction, and a variable arrangement of bridging leaflets that may form one shared valve or two tethered valves. The structural consequences are always a combination of a primum atrial septal defect, an inlet ventricular septal defect, and abnormalities of the left atrioventricular valve, traditionally called a cleft mitral valve. The exact mix of these determines where a patient sits on the spectrum. [1] [2]
The reason this lesion earns a high-yield place in every fellowship curriculum is that it sits at the intersection of anatomy, genetics, and surgical timing. It is the commonest major cardiac lesion in Down syndrome, it is one of the few congenital defects with a signature ECG finding, and it has a hard deadline — repair the complete form early or lose the patient to pulmonary vascular disease. Hold those three threads together, and the rest of the topic falls into place around them. [3] [7]
Classification
The classification question examiners love is the spectrum from partial to complete, because it predicts everything downstream — presentation, pulmonary pressures, and timing of surgery. The three forms share the same deficient atrioventricular septum but differ in how the bridging leaflets are arranged and whether there is a ventricular component to the shunt. [1]
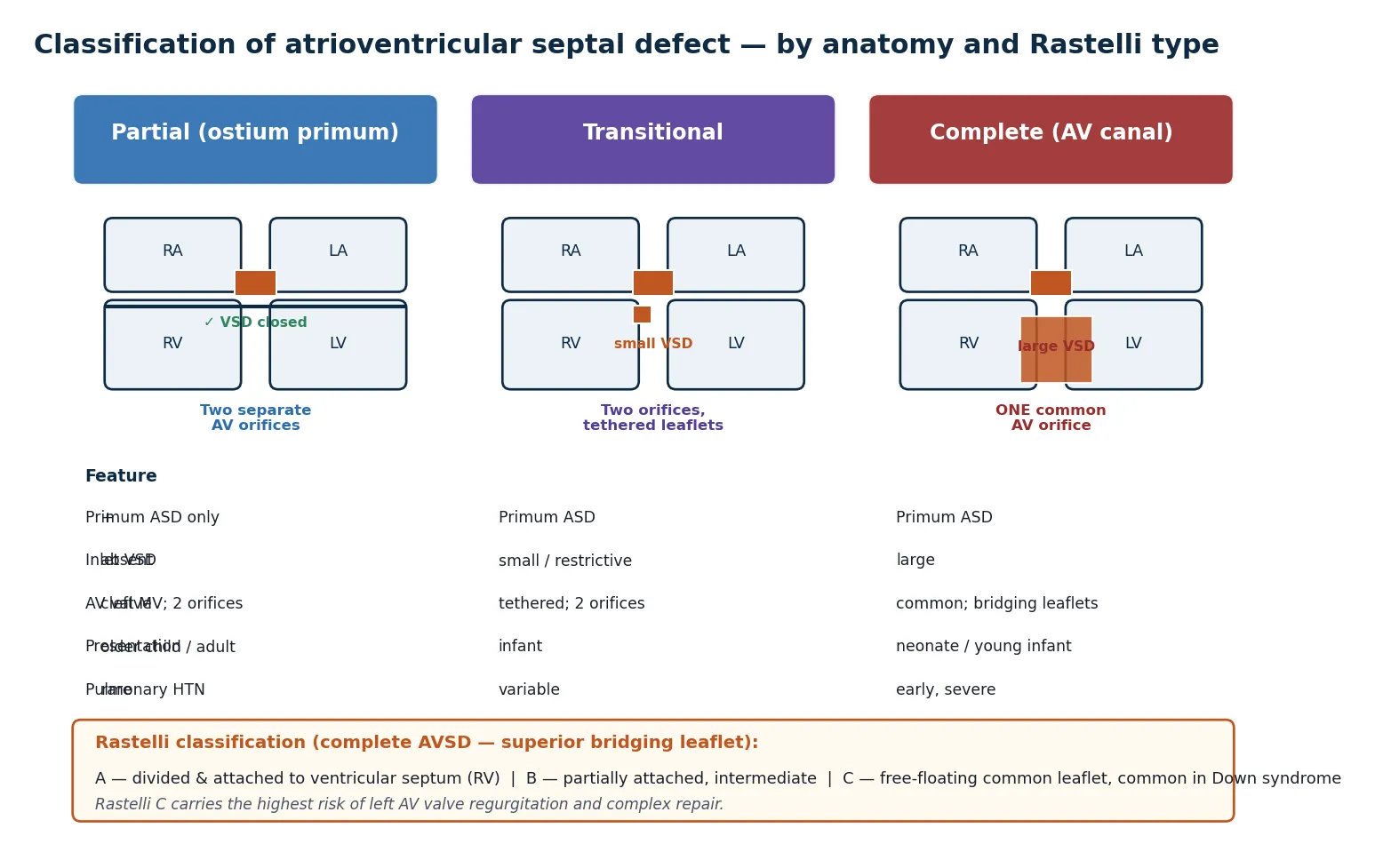
The partial form consists of an ostium primum atrial septal defect with a cleft in the anterior leaflet of the mitral valve and no ventricular septal defect. There are two separate atrioventricular orifices, so the shunt is confined to the atrial level. These children present later, often as older infants or even adults, with a murmur, recurrent chest infections, or an incidental finding, and they rarely develop pulmonary hypertension in childhood. [1]
The transitional form sits between the two. It has a primum atrial defect, two atrioventricular orifices, and a small, usually restrictive inlet ventricular septal defect produced by the leaflets being tethered down to the septum by short chordae. The clinical behaviour is closer to the partial form, but the presence of any ventricular communication raises the pressure load on the right ventricle. [2]
The complete form is the classic atrioventricular canal. There is a large primum atrial septal defect, a large non-restrictive inlet ventricular septal defect, and a single common atrioventricular valve with superior and inferior bridging leaflets spanning both ventricles. These infants present in the first weeks of life with heart failure and develop pulmonary hypertension early, which is why repair is scheduled before the pulmonary vasculature is irreversibly damaged. [2] [7]
The complete form is further graded by the Rastelli classification, which describes how the superior bridging leaflet is tethered. In Rastelli type A the leaflet is divided and attached to the crest of the ventricular septum; in type B it attaches anomalously to an anomalous papillary muscle on the right side; and in type C the leaflet is free-floating and undivided, tethered only to the anterior papillary muscle of the right ventricle. Rastelli C is the form most often seen in Down syndrome and carries the highest risk of left atrioventricular valve regurgitation and a more complex repair. [2]
Epidemiology & Risk Factors
AVSD accounts for around three to five percent of all congenital heart defects, but the headline number hides the strength of its genetic signature. The single most important risk factor is Down syndrome. Roughly one in two children with trisomy 21 has a congenital heart defect, and of those, the atrioventricular septal defect — particularly the complete form — is the lesion most strongly and specifically associated, far more than ventricular or atrial septal defects seen in the general population. [3] [4]
The reverse statistic is the one examiners probe. Of all children born with a complete AVSD, up to three quarters have Down syndrome. This bidirectional link is so reliable that a fetus or infant with a complete canal defect should be assumed to carry trisomy 21 until karyotyping confirms otherwise, and a prenatal diagnosis of AVSD is itself an indication for genetic testing of the pregnancy. [3] [5]
Beyond trisomy 21, AVSD recurs in a handful of less common syndromes and chromosomal disorders. It appears in heterotaxy syndromes, in Smith-Lemli-Opitz and Ellis–van Creveld syndromes, and as part of the cardiac phenotype of several single-gene disorders such as Noonan and Holt-Oram. Maternal diabetes and pregestational diabetes modestly raise the risk of endocardial cushion defects, and the recurrence risk for siblings of an affected child in the absence of a syndrome is around three percent, rising if a parent carries a balanced translocation. [4] [5]
Pathophysiology
To understand why this lesion behaves the way it does, trace the blood through the shared junction. The deficient atrioventricular septum leaves two holes — a primum atrial defect above the valve and an inlet ventricular defect below it — and the malformed common valve sits across both. Blood takes the path of least resistance, shunting left to right at both levels because the right ventricle and pulmonary circulation are the lower-pressure side. The result is volume overload of the right heart and massive pulmonary overcirculation. [1] [2]
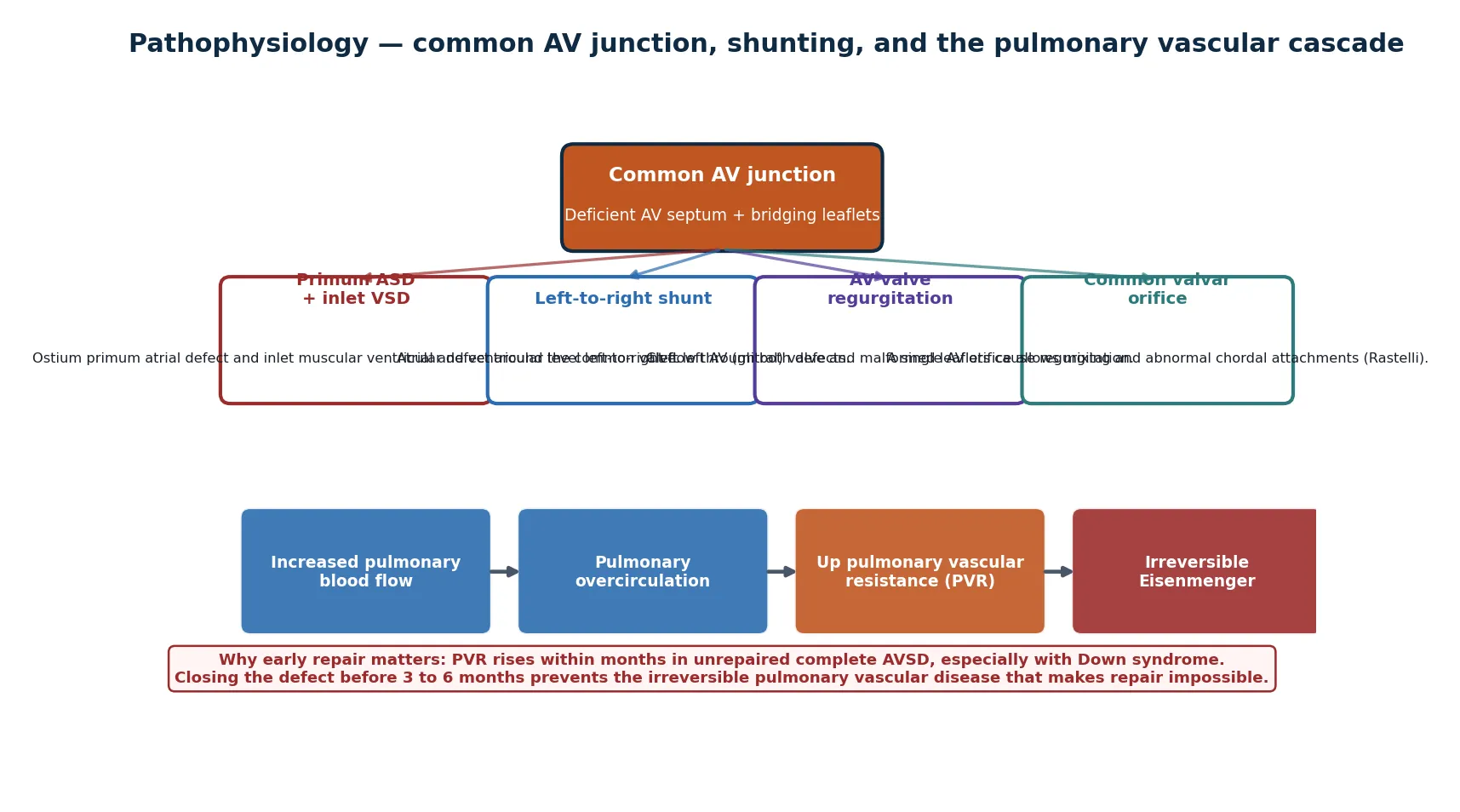
The magnitude of the shunt depends on the form. In a partial defect the shunt is atrial and the pulmonary-to-systemic flow ratio is moderate, so the right ventricle copes for years. In a complete defect the large inlet VSD makes the shunt ventricular as well as atrial, and the common valve allows free mixing, so pulmonary blood flow is several times systemic. That is why the complete form causes congestive heart failure in the first weeks of life while the partial form may be silent into adulthood. [2]
The second engine of trouble is the malformed valve. The left atrioventricular valve in AVSD is anatomically different from a normal mitral valve — it has three leaflets, the so-called cleft, and its tension apparatus is abnormal. This produces the left AV valve regurgitation that is the dominant long-term problem after repair and the leading reason patients come back to theatre. The cleft is not a defect to be dismissed as cosmetic; it is the central issue of surgical planning. [9]
The pulmonary vasculature is where the pathology turns dangerous. Sustained high pulmonary blood flow remodels the small pulmonary arteries, raising pulmonary vascular resistance. In a complete AVSD this process accelerates, and the media and intima of the pulmonary arterioles thicken irreversibly within the first months of life — faster still in Down syndrome, where the pulmonary vasculature is intrinsically more reactive. Once resistance exceeds systemic, the shunt reverses and Eisenmenger physiology closes the door on surgical repair forever. [7]
The five components of a complete AVSD — CRUX
Clinical Presentation
The presentation is dictated by the form and therefore by the age at which the child is seen. A complete AVSD declares itself in early infancy with the familiar picture of a large left-to-right shunt — tachypnoea, sweating with feeds, poor weight gain, and recurrent lower respiratory tract infections. The mother often describes exhausting feeds and a baby who is working harder to breathe than she remembers. By two to three months, untreated complete defects have declared themselves in failure to thrive. [2]
On examination the clues accumulate. There is a hyperactive precordium from the volume-loaded right ventricle, a systolic thrill at the lower left sternal edge, and a pansystolic murmur of atrioventricular valve regurgitation. The pulmonary flow murmur is heard as a systolic ejection murmur at the upper left sternal edge, and a mid-diastolic rumble at the apex reflects increased flow across the mitral valve. The second heart sound is usually narrowly split with a loud pulmonary component once pulmonary hypertension develops. [1]
A partial AVSD is the great imitator. Because the shunt is atrial and moderate, these children may present late — sometimes into adulthood — with the incidental finding of a murmur, exercise intolerance, or an unusual chest infection. The trap is mistaking it for a secundum atrial septal defect. The two share a right ventricular heave and a pulmonary flow murmur, but the partial AVSD adds the cleft mitral valve and its regurgitant murmur, and the ECG tells them apart instantly. [1]
Increasingly the first presentation is antenatal. Fetal echocardiography detects the complete form in the second trimester, and a suspected AVSD on the four-chamber view — with a common AV valve and a defect at the crux — prompts karyotyping and detailed counselling about the postnatal plan. Prenatal detection shifts the entire pathway upstream, allowing planned delivery in a cardiac centre and early surgical referral. [8]
Differential Diagnosis
The differential turns on the level of the shunt and the presence or absence of pulmonary hypertension. The complete AVSD with neonatal heart failure sits alongside the other causes of a large left-to-right shunt presenting in infancy, while the partial form masquerades as a simple atrial septal defect. The ECG is the single fastest discriminator. [1]
A large ventricular septal defect produces a similar picture of infant heart failure with a pansystolic murmur, but the ECG shows a normal or leftwards axis rather than the superior axis of AVSD, and echocardiography localises the defect to the membranous or muscular septum with a normal crux. An ostium secundum ASD, the chief mimic of the partial form, has a normal axis and a structurally normal atrioventricular valve. [1]
A truncus arteriosus and an atrioventricular defect with a single great vessel can both cause early heart failure and pulmonary overcirculation, but truncus has a single semilunar valve and overriding arterial trunk on imaging. A double-inlet left ventricle or an unbalanced AVSD with a hypoplastic ventricle may be confused with a hypoplastic left heart syndrome, and these need detailed echocardiography to separate a two-ventricle repair from a single-ventricle Fontan pathway. [2]
Clinical & Bedside Assessment
The bedside assessment is where the fellowship candidate earns marks, because the physical signs are distinctive and the ECG finding is pathognomonic. Begin with growth, work of breathing, and perfusion, since these place the child on the severity spectrum before you ever touch the stethoscope. [1]
Inspection reveals a child who may be tachypnoeic at rest, with subcostal recession and a hyperactive precordium visible through the chest wall. Palpation confirms a systolic thrill at the lower left sternal edge in the complete form and a right ventricular heave from the volume load. The apex is thrusting and displaced when the left AV valve regurgitation is significant. Hepatomegaly reflects right heart congestion in advanced disease. [1]
Auscultation is interpreted in layers. The first heart sound may be single and loud from the common valve. A pansystolic murmur at the lower left sternal edge reflects the AV valve regurgitation, a systolic ejection murmur at the upper left sternal edge reflects increased pulmonary flow, and a mid-diastolic rumble at the apex reflects increased flow across the left AV valve. The second heart sound is widely split but, critically, the pulmonary component becomes loud and delayed as pulmonary hypertension develops. [1]
The investigation that changes everything is the ECG, and it should be obtained on every child with a suspected atrial level shunt. The signature finding is a superior QRS axis, typically between −30 and −150 degrees, produced by the abnormal inferior-to-superior activation of the ventricles through the deficient septum. This is the single feature that distinguishes a primum from a secundum ASD at the bedside, and examiners test it relentlessly. Right atrial enlargement, right ventricular hypertrophy, and a prolonged PR interval are the associated findings. [1] [2]
Investigations
Echocardiography is the definitive investigation and usually the only one needed before surgery. A complete segmental study defines the anatomy — the primum ASD, the inlet VSD, the arrangement of the bridging leaflets, the Rastelli type, and the competence of the left atrioventricular valve — and it assesses ventricular balance, the size and function of both ventricles, and the presence of associated lesions such as a persistent left superior vena cava or coarctation. Colour and continuous-wave Doppler estimate the pulmonary artery pressure from any tricuspid regurgitation jet. [1] [8]
The chest radiograph supports but never diagnoses. It shows cardiomegaly with right atrial and right ventricular enlargement, increased pulmonary vascular markings from the left-to-right shunt, and, in older unoperated cases, pruning of the peripheral vessels as pulmonary vascular disease takes hold. It is most useful for tracking heart failure over time and for excluding alternative causes of respiratory distress. [2]
Cardiac catheterisation is reserved for the difficult decisions. It is used to measure pulmonary vascular resistance directly when operability is in question, particularly in the older infant or child presenting late, or in the adult with a partial defect and suspected Eisenmenger physiology. The haemodynamic question is whether the pulmonary vascular resistance falls with vasodilator testing, because a fixed, high resistance rules out repair. [7] [10]
Genetic testing is part of the work-up, not an afterthought. Karyotype or chromosomal microarray confirms or excludes Down syndrome and identifies the rarer syndromic associations, which carry implications for counselling, recurrence risk, and the long-term management plan. Cardiac genetics referral is appropriate for every child with an AVSD and a dysmorphic phenotype or a family history of congenital heart disease. [4] [5]
Management — Resuscitation

The resuscitation phase applies almost entirely to the complete form presenting in heart failure. The infant who is tachypnoeic, failing to thrive, and sweating with feeds needs medical stabilisation to grow and reach a safe weight for surgery, but never at the cost of delaying the definitive repair past the pulmonary vascular deadline. [2]
Diuretics are the cornerstone of medical therapy. Furosemide, with or without spironolactone or amiloride, reduces pulmonary congestion and eases the work of breathing, allowing the infant to feed and gain weight. Fluid restriction and caloric supplementation — fortified expressed breast milk or high-energy formula — address the failure to thrive that is the clinical signature of the large shunt. [1]
An angiotensin-converting enzyme inhibitor such as captopril or enalapril is added when left atrioventricular valve regurgitation is significant, reducing the afterload on the left ventricle and the volume of regurgitant flow. Digoxin has a role in infants with a tachyarrhythmia or marked ventricular dysfunction but is not first-line for the pure volume overload. Pulmonary vasodilators such as bosentan or sildenafil have no routine place before repair and are reserved for the child found to be inoperable. [2] [7]
Furosemide
Dose
0.5–1 mg/kg/dose orally every 8–12 hours
The decisive action is not medical but logistical. The moment a complete AVSD is confirmed, the surgical referral is made and a repair date is set within the first three to six months. Medical therapy buys time and weight; it does not buy a longer pulmonary vascular window. Every month of delay in a complete defect is a month of irreversible remodelling of the pulmonary arterioles. [7]
Management — Definitive & Stepwise
Definitive management is surgical, and the timing and technique are determined by the form. The principle that governs the whole pathway is early and complete closure of the septal defects with reconstruction of a competent left atrioventricular valve, performed before pulmonary vascular disease becomes irreversible. [1]
For the complete form, primary surgical repair is performed at three to six months of age, before pulmonary vascular resistance has climbed irreversibly. The operation closes both the primum atrial and the inlet ventricular defects and divides the common valve into two competent orifices. Two techniques are used — the single-patch (or modified single-patch / nunn) technique apposes the bridging leaflets directly to the crest of the septum, while the two-patch technique uses separate patches for the atrial and ventricular defects. The modified single-patch has gained favour for its shorter, simpler operation with comparable outcomes in suitable anatomy. [2] [6]
For the partial form, repair is elective and usually performed in early childhood, since the slower course gives more latitude. The operation closes the primum defect and repairs the cleft in the left AV valve, the latter being the technical detail that determines late valve function. The decision to repair the cleft fully or partially balances the risk of stenosis against the risk of residual regurgitation. [1]
Surgical decision sequence
Confirm anatomy on echocardiography — form, Rastelli type, ventricular balance, AV valve competence
Assess pulmonary vascular resistance if presenting late or any doubt about operability
Complete form — book primary repair at 3–6 months; partial form — elective repair in early childhood
Choose single-patch vs two-patch by anatomy; reconstruct competent left AV valve
Postoperative surveillance for residual VSD, AV valve regurgitation, and left ventricular outflow obstruction
An unbalanced AVSD, where one ventricle is hypoplastic, cannot be repaired into two ventricles and follows the single-ventricle pathway — pulmonary artery banding or staged palliation culminating in a Fontan circulation. Recognising the unbalanced form early prevents an futile attempt at biventricular repair and sets the family on the correct, though longer, surgical road. [2]
Specific Subtypes & Scenarios
AVSD in Down syndrome is the scenario most often examined because it brings together genetics, anatomy, and surgical outcomes. Roughly half of children with trisomy 21 have a cardiac defect, and the AVSD — particularly the complete form — is over-represented far beyond chance. The good news, and the reassurance families need, is that surgical outcomes in Down syndrome are excellent and comparable to the non-syndromic population when repair is timely, so trisomy 21 is never a reason to defer surgery. The caution is that the pulmonary vasculature in Down syndrome is more reactive, so the early-repair principle applies with even greater force. [3] [7]
Prenatally detected AVSD has transformed the pathway. Fetal echocardiography identifies the complete form on the four-chamber view, showing the common atrioventricular valve and the defect at the crux, and this triggers karyotyping, counselling about trisomy 21, and planning for delivery in a cardiac centre. The partial form is harder to detect antenatally but is increasingly picked up with improved imaging. Prenatal diagnosis allows planned postnatal care and avoids the late presentation that compromises the pulmonary vascular window. [8]
Late presentation in the older child or adult almost always means a partial AVSD that was missed. These patients present with a murmur, exercise intolerance, atrial arrhythmia, or an incidental finding, and the question that dominates their assessment is whether the pulmonary vascular resistance is still operable. Cardiac catheterisation is essential here, and a fixed high resistance converts the management from surgical repair to medical management of pulmonary hypertension. [10]
The unbalanced AVSD is the form that breaks the standard pathway. When one ventricle is too small to support the circulation, biventricular repair is impossible and the child is committed to the single-ventricle Fontan pathway, staged over the first few years of life. Early recognition by echocardiography — measuring ventricular volumes and the degree of imbalance — prevents an unsuccessful attempt at two-ventricle repair and sets realistic expectations. [2]
Complications & Pitfalls
The complications divide into those of unrepaired disease and those of the repaired heart. The gravest complication of unrepaired complete AVSD is irreversible pulmonary vascular disease, progressing to Eisenmenger physiology with cyanosis, pulmonary hypertension, and eventual right heart failure. Once the shunt has reversed, surgery is no longer an option, and management shifts to pulmonary vasodilator therapy and the management of the complications of chronic cyanosis. [7] [10]
After repair, the dominant problem is left atrioventricular valve regurgitation. Despite meticulous cleft closure, a proportion of patients develop progressive regurgitation of the reconstructed valve, and this is the leading reason for reoperation over the long term. Residual or recurrent ventricular septal defect, left ventricular outflow tract obstruction from the elongated outflow that AVSD anatomy produces, and complete heart block from sutures near the conduction tissue are the other recognised postoperative issues. [9]
The conduction system is at risk because the atrioventricular node is displaced inferiorly and posteriorly in AVSD, sitting in an abnormal position relative to the surgical patches. Transient heart block is common postoperatively; permanent complete heart block requiring pacing is uncommon but serious. Postoperative arrhythmias, including junctional ectopic tachycardia, demand vigilance in the immediate postoperative period. [2]
The pitfall that catches candidates is treating an AVSD as if it were a simple ASD. The partial form in particular invites the assumption that it behaves like a secundum defect, but the cleft mitral valve, the superior axis, and the different surgical approach all distinguish it. Another trap is delaying referral of a complete defect for medical optimisation past the six-month window, sacrificing operability for weight gain that does not change the pulmonary vasculature. [1] [7]
Long-term complications after AVSD repair
Prognosis & Disposition
The prognosis of a repaired AVSD is excellent and is one of the success stories of congenital cardiac surgery. Early mortality for primary repair of complete AVSD in modern centres is now well under five percent, and the vast majority of children grow up with normal or near-normal exercise capacity and quality of life. The partial form carries even lower operative risk, though its longer latent course means presentation may be late. The key determinant of a good outcome is timely repair before pulmonary vascular disease. [6] [9]
Long-term survival is dominated by the fate of the left atrioventricular valve. Reoperation for valve regurgitation occurs in roughly ten to fifteen percent of patients over the long term, and a small proportion ultimately require valve replacement. Patients who escape reoperation and significant residual lesions can expect a near-normal life expectancy, though they remain under lifelong surveillance. [9]
Disposition after repair is to lifelong follow-up in a specialist congenital cardiac service. The repaired AVSD is never "cured" in the sense that surveillance can stop — residual lesions, valve dysfunction, arrhythmia, and the rare late pulmonary hypertensive crisis all demand ongoing review. Transition to an adult congenital heart disease service in late adolescence is essential, with specific counselling about pregnancy, exercise, and endocarditis prophylaxis. [10]
For the child found to be inoperable because of irreversible pulmonary vascular disease, the prognosis is fundamentally different. Management is medical, centred on pulmonary vasodilator therapy, management of the complications of chronic cyanosis, and avoidance of factors that worsen pulmonary hypertension. These patients require specialist pulmonary hypertension care and carry a markedly reduced life expectancy. [7] [10]
Special Populations
Down syndrome is the population in which AVSD is most often encountered and in whom the management principles are most stringent. The association is so strong that a complete AVSD is trisomy 21 until proven otherwise, and the pulmonary vasculature is more reactive, so early repair is non-negotiable. Reassure families that surgical outcomes are excellent and comparable to non-syndromic children, and address the broader developmental and medical needs of the child with Down syndrome through a coordinated multidisciplinary approach. [3] [4]
Migrant and refugee families may present late with an unrecognised complete or partial AVSD, having had limited antenatal and postnatal cardiac screening. The challenge here is twofold — the pulmonary vascular window may already be closing, and the family may face language, cultural, and healthcare-access barriers. A planned assessment of operability, interpreter-mediated counselling, and clear coordination with surgical and social services determine the outcome. [10]
Adolescents and young adults with repaired AVSD face the transition from paediatric to adult congenital cardiac care, a handover that is often poorly executed and carries real risk. The issues are the fate of the left AV valve, the emergence of atrial arrhythmia, left ventricular outflow tract obstruction, and the counselling needed around pregnancy, contraception, and the heritability of congenital heart disease. A structured transition programme in late adolescence is the standard of care. [10]
Evidence, Guidelines & Regional Differences
The evidence base for AVSD management is built on decades of surgical outcome data rather than randomised trials, reflecting the nature of congenital cardiac surgery. The Society of Thoracic Surgeons congenital database and equivalent registries underpin the patterns of practice and outcomes that guide timing and technique, documenting the steady decline in operative mortality and the centrality of the left AV valve to long-term results. [6] [9]
Guidelines converge on the same principles. The European Society of Cardiology guidelines for the management of adult congenital heart disease set the framework for long-term follow-up, pregnancy counselling, and the management of residual lesions, while the American Heart Association scientific statements on the genetic basis of congenital heart disease inform the work-up and counselling of the family. Both emphasise lifelong surveillance and structured transition. [5] [10]
In Australia and New Zealand, antenatal detection rates are high, and complete AVSDs are routinely referred for surgical repair at three to six months through the specialist paediatric cardiac centres. Long-term follow-up is coordinated through adult congenital cardiac services, with particular attention to the needs of rural and remote families and Aboriginal and Torres Strait Islander children, for whom access and continuity of care are ongoing priorities. [3]
Regional differences matter most where access to fetal and paediatric echocardiography is limited. In resource-constrained settings, late presentation of complete AVSD with established pulmonary vascular disease is more common, and the operability question dominates management. Telemedicine links to specialist centres, training in point-of-care cardiac assessment, and clear referral pathways are the practical responses. The underlying principle — that operability is a function of pulmonary vascular resistance and therefore of time — does not change with geography. [7] [10]
Exam Pearls
When the examiner hands you an AVSD, lead with the anatomy at the crux and the superior axis, and the rest follows. The hierarchy of high-yield facts is consistent across every exam cycle: the common junction, the partial–transitional–complete spectrum, the Rastelli classification, the Down syndrome association, the superior QRS axis, the three-to-six-month surgical window, and the left atrioventricular valve as the long-term problem. [1] [2]
AVSD exam anchors
The viva traps are predictable. Expect to be asked to distinguish a partial AVSD from a secundum ASD — the answer is the superior axis and the cleft valve. Expect a question on why the complete form is repaired early — the pulmonary vascular cascade and Eisenmenger physiology. Expect to be asked about the unbalanced form and why it cannot be repaired with two ventricles. And expect a long-case discussion of a Down syndrome infant with a complete canal, where the counselling, the surgical plan, and the prognosis are all testable. [1] [3]
Society of Thoracic Surgeons Congenital Heart Surgery Database — AVSD outcomes
Key finding
Multi-institutional registry data show operative mortality for complete AVSD repair below 5% in modern practice, with left AV valve regurgitation the dominant driver of reoperation (~10–15% long-term). Early primary repair before 6 months prevents irreversible pulmonary vascular disease.
References
- [1]Craig B Atrioventricular septal defect: from fetus to adult. Heart, 2006.PMID 17105897
- [2]Calabrò R, Limongelli G Complete atrioventricular canal. Orphanet J Rare Dis, 2006.PMID 16722604
- [3]Bergström S, Carr H, Petersson G, et al Trends in congenital heart defects in infants with Down syndrome. Pediatrics, 2016.PMID 27252035
- [4]Pierpont ME, Basson CT, Cyran SE, et al Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association. Circulation, 2007.PMID 17519398
- [5]Pierpont ME, Brueckner M, Chung WK, et al Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation, 2018.PMID 30571578
- [6]Jacobs JP, O'Brien SM, Pasquali SK, et al Atrioventricular septal defects: lessons learned about patterns of practice and outcomes from the congenital heart surgery database of the society of thoracic surgeons. World J Pediatr Congenit Heart Surg, 2010.PMID 23804725
- [7]Suzuki K, Yamaki S, Mimori S, et al Pulmonary vascular disease in Down's syndrome with complete atrioventricular septal defect. Am J Cardiol, 2000.PMID 10946038
- [8]Paladini D, Marasini M, Volpe P, et al Partial atrioventricular septal defect in the fetus: diagnostic features and associations. Ultrasound Obstet Gynecol, 2009.PMID 19705406
- [9]O'Connor M, Bellsham-Revell H, Pushparajah K, et al The fate of the left atrioventricular valve after atrioventricular septal defect repair. Pediatr Cardiol, 2026.PMID 40208292
- [10]Baumgartner H, De Backer J, Babu-Narayan SV, et al The ESC clinical practice guidelines for the management of adult congenital heart disease. Eur Heart J, 2020.PMID 33128054