Paeds · cardiology
Cardiomyopathies in children
Also known as Paediatric cardiomyopathy · Hypertrophic cardiomyopathy in children · Dilated cardiomyopathy in children · Restrictive cardiomyopathy in children · Left ventricular non-compaction
Fellowship guide to the paediatric cardiomyopathies: the morphological framework (dilated, hypertrophic, restrictive, non-compaction, arrhythmogenic), the genetic substrate and cascade family screening, the pathophysiology that drives heart failure, arrhythmia and sudden cardiac death, the bedside and echocardiographic assessment, the medical, device and transplant management, and the long-term prognosis that makes this the commonest reason for paediatric heart transplantation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A cardiomyopathy is a disease of the heart muscle in which the abnormality of structure or function is intrinsic to the myocardium itself, not the secondary consequence of hypertension, valve disease, or congenital obstruction. The 2006 American Heart Association scientific statement defined the cardiomyopathies as "a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction", and the 2008 European Society of Cardiology position statement framed the same idea operationally: classify by morphology first, then ask what caused it. Together these two documents are the frame every fellowship candidate must reproduce when asked "what is a cardiomyopathy?" [2] [3]
In children the cardiomyopathies behave very differently from the adult disease. They are uncommon — the population incidence is roughly one per hundred thousand children per year — yet they are the single largest diagnosis group leading to paediatric heart transplantation, and sudden cardiac death in the young is dominated by occult cardiomyopathy. The clinical problem they pose is therefore not the volume of cases but the gravity of each one: a missed diagnosis is a child who collapses on the sports field, or an infant who presents in fulminant heart failure. [1] [6]
Classification
The cardiomyopathies are classified first by morphology — what the echocardiogram shows — because morphology drives the immediate differential, the symptoms, and the complication to watch for. The genotype then refines the risk: which relatives to screen, the probability of sudden death, and the surveillance interval. A candidate who states only the morphology, or only the gene, has given half the answer. [2] [3]
The five morphological types are the working set. Dilated cardiomyopathy (DCM) is a dilated, thin-walled left ventricle with impaired systolic function; it is the commonest type in children and the commonest cause of paediatric heart failure. Hypertrophic cardiomyopathy (HCM) is an inappropriately thick, often small-cavity ventricle with preserved or hyperdynamic systolic function but impaired filling; it is the cardiomyopathy most associated with sudden cardiac death in the young. Restrictive cardiomyopathy (RCM) is a stiff, poorly filling ventricle with markedly enlarged atria; it is rare but carries the worst prognosis. Left ventricular non-compaction (LVNC) is characterised by prominent trabeculations and deep intertrabecular recesses; its classification as a distinct cardiomyopathy is debated but it is managed as one. Arrhythmogenic cardiomyopathy (ACM, formerly arrhythmogenic right ventricular cardiomyopathy) is a progressive fibro-fatty replacement of myocardium that presents predominantly with arrhythmia rather than failure. [2] [3] [10]

The genetic layer is now inseparable from the classification. A pathogenic variant can be identified in roughly thirty to forty percent of children with a cardiomyopathy, with the highest yield in HCM. Most childhood HCM is autosomal dominant and caused by sarcomeric protein mutations, classically in MYH7 and MYBPC3. Familial DCM is also frequently genetic, with cytoskeletal, sarcomeric, and nuclear-envelope genes implicated. Restrictive disease, LVNC, and ACM each have their own gene panels. The practical consequence is that every child with a cardiomyopathy deserves genetic testing, and every first-degree relative of a genotype-positive case deserves cascade screening. [4] [8]
Epidemiology & Risk Factors
The population incidence of paediatric cardiomyopathy was established by the Pediatric Cardiomyopathy Registry in the United States. Lipshultz and colleagues reported an incidence of approximately 1.13 cases per 100 000 children per year across two regions, with dilated cardiomyopathy the commonest type in infancy and hypertrophic cardiomyopathy more evenly distributed across age. The incidence is higher in the first year of life than at any other age, and higher in boys than girls for most types. These figures are the canonical reference an examiner expects when the question turns to "how common is it?" [1]
The Australian national population studies provide the outcomes counterpart. Alexander and colleagues' study of hypertrophic cardiomyopathy diagnosed in childhood, and Shi and colleagues' study of childhood left ventricular non-compaction, both drew on the same national cohort that tracks every Australian child with a cardiomyopathy. They documented the long-term freedom from death and transplant and the competing risks of sudden death and progressive failure. These studies are especially important in the ANZ context because they give local, population-based outcome data rather than tertiary-centre referral bias. [9] [10]
The risk factors are overwhelmingly genetic. A first-degree family history of cardiomyopathy or unexplained sudden death before the age of fifty is the single most important predictor of disease in a child, and it is the question that most often unearths an inherited cardiac condition. Inborn errors of metabolism, syndromic associations (Noonan and related RASopathies for HCM; Barth syndrome and mitochondrial disease for DCM and LVNC), prior viral myocarditis, anthracycline chemotherapy, and chronic tachyarrhythmia are the principal acquired and secondary causes of DCM in children. The counselling point is that a new diagnosis of cardiomyopathy in a child is a diagnosis for the family: every first-degree relative needs evaluation. [4] [8]
Pathophysiology
The cardiomyopathies share a final common mechanism: an intrinsic myocardial abnormality that progressively degrades the heart's pump function and its electrical stability. The details differ by morphology, which is why the type matters. In dilated cardiomyopathy the abnormal cytoskeleton and sarcomere cannot maintain myocyte integrity under load, the wall stress rises, the ventricle remodels and dilates, and the ejection fraction falls. The dominant physiology is systolic heart failure — poor output, compensatory neurohormonal activation, fluid retention, and growth failure in the infant. [8]
In hypertrophic cardiomyopathy a sarcomeric mutation produces myocyte disarray and hypertrophy, the ventricle becomes thick and small, and the problem shifts from contraction to filling: diastolic dysfunction raises filling pressures and produces exertional symptoms. A subset develop dynamic left ventricular outflow tract obstruction, and the disarrayed myocytes with intervening fibrosis create the arrhythmogenic substrate that drives sudden cardiac death. Maron's systematic review established the scale of the sudden-death problem in HCM, and the randomised evidence for implantable defibrillators in high-risk patients came from Maron and colleagues' landmark study. [4] [5]
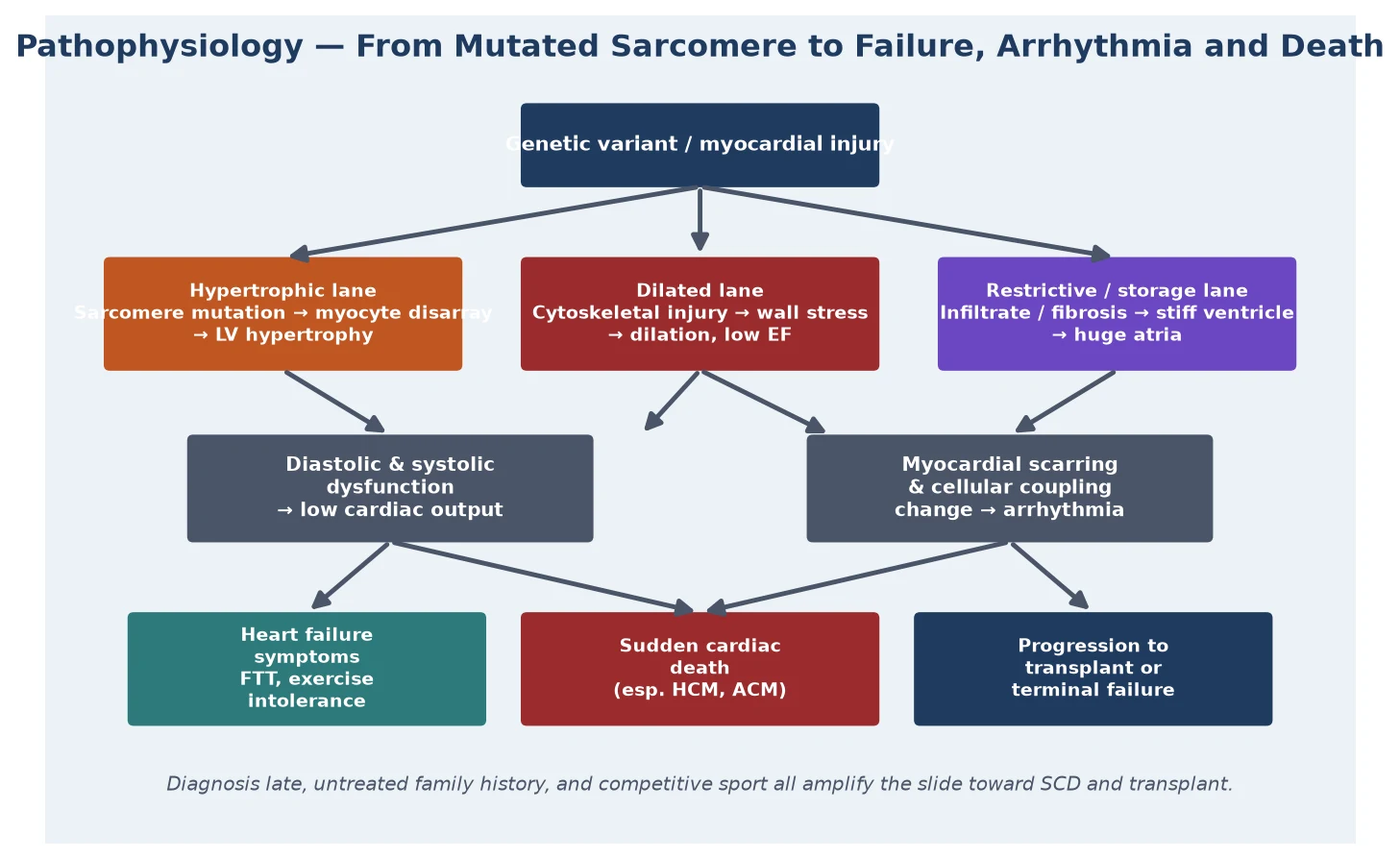
Restrictive cardiomyopathy is the pathophysiological opposite of dilated: the ventricle is not weak but stiff, with poor compliance that drives filling pressures sky-high and balloons the atria. The result is biventricular inflow obstruction at the atrial level, low preload to the ventricles, and a child who is at once at risk of heart failure and of sudden death from the same fibrotic, infiltrative process. Left ventricular non-compaction reflects a failure of myocardial compaction in development, leaving a trabeculated, spongy wall that impairs systolic function and predisposes to thromboembolism and arrhythmia. Arrhythmogenic cardiomyopathy replaces myocardium with fibro-fatty tissue, usually beginning in the right ventricle, and presents predominantly as arrhythmia. [3] [10]
Clinical Presentation
The presentation depends on the type, the age, and whether the diagnosis was made by screening a family or by symptoms. Many children are now diagnosed before symptoms because a relative has been found to carry a pathogenic variant; these screened children may be entirely well, and the task is surveillance rather than treatment. Symptomatic presentation is the more dangerous pathway because it means the disease has already declared itself. [1] [4]
An infant with dilated cardiomyopathy presents with heart failure: poor feeding, sweating with feeds, tachypnoea, failure to thrive, recurrent respiratory infections, hepatomegaly, and a gallop rhythm. An older child with DCM presents with exercise intolerance, abdominal pain, and the signs of biventricular failure. A child with hypertrophic cardiomyopathy may present asymptomatically with a murmur, or symptomatically with exertional chest pain, breathlessness, pre-syncope, syncope, or — at the catastrophic end — sudden cardiac death on the sports field. Restrictive cardiomyopathy presents with right-heart failure, hepatomegaly, ascites, and exertional symptoms. LVNC and ACM present variably with heart failure, arrhythmia, thromboembolism, or sudden death. [4] [9]
The family history is part of the presentation, not an afterthought. A history of sudden unexplained death before the age of fifty, of "heart attacks" in young relatives, of deafness and arrhythmia, or of a known cardiomyopathy in a parent reframes an otherwise vague story into a high-risk evaluation. Equally, a child who presents with cardiomyopathy must trigger a three-generation family history, because the diagnosis is often familial and the relatives are at risk. [4] [8]
Differential Diagnosis
The differential splits into two questions: is this really a primary cardiomyopathy, and if so which type? The first question matters because several conditions mimic cardiomyopathy and have entirely different management. Untreated supraventricular tachycardia can produce a tachycardiomyopathy that is fully reversible with rate or rhythm control; this must be excluded in any infant with DCM and a fast heart rate. Viral myocarditis can present identically to acute DCM and the two are often indistinguishable at first; the history, viral studies, and occasionally magnetic resonance imaging help separate them. Anthracycline cardiotoxicity, thyrotoxicosis, sepsis, and severe anaemia can all depress function and must be considered and excluded. [7] [8]
The second question — which type — is resolved by echocardiography. The athlete's heart is the classic mimic of HCM: physiological remodelling from intensive training can thicken the wall and overlap morphologically with early HCM. The discriminators are the degree of hypertrophy, the pattern (asymmetric septal in HCM), diastolic function, the electrocardiogram, and crucially a family history and genetic testing. A child with apparent RCM must have infiltrative and storage disease — particularly the inherited metabolic and lysosomal storage disorders — sought and excluded, because some are treatable. [3] [9]
Clinical & Bedside Assessment
Bedside assessment rests on three pillars: the history (symptoms and family), the examination (failure signs and murmur), and the resting electrocardiogram. The history probes exertional symptoms — chest pain, breathlessness, palpitations, syncope — and the feeding and growth pattern in infants, then constructs a three-generation family tree focused on sudden death, cardiomyopathy, pacemakers or defibrillators, and "heart attacks" in the young. The family history is not optional: it is the single highest-yield piece of information in the evaluation. [4] [6]
Examination looks for the signs of heart failure — tachypnoea, tachycardia, hepatomegaly, gallop, poor perfusion, failure to thrive — and for the morphological clue. An ejection systolic murmur at the left sternal border that increases with reducing preload (standing or Valsalva) suggests dynamic LVOT obstruction in HCM. A loud apical holosystolic murmur suggests mitral regurgitation in DCM. A right ventricular heave, raised jugular venous pressure, ascites, and hepatomegaly suggest restrictive physiology. However, examination can be entirely normal even in significant disease, which is why a normal examination never excludes a cardiomyopathy when the history or ECG is abnormal. [2] [4]
The resting twelve-lead electrocardiogram is the third pillar and must never be omitted. In HCM it is almost always abnormal, with voltage criteria for left ventricular hypertrophy, repolarisation abnormalities, deep T-wave inversions, and pathological Q waves. In DCM it may show left ventricular hypertrophy, flat or inverted T waves, or conduction disease. A completely normal ECG makes HCM highly unlikely and should prompt reconsideration of an athlete's-heart or innocent-murmur diagnosis. The ECG is also the gateway to detecting the arrhythmia that may herald sudden death. [4] [9]
Investigations
Echocardiography is the cornerstone investigation and is mandatory in any child with suspected cardiomyopathy. It defines the morphology — chamber sizes, wall thickness, ejection fraction, and the pattern of hypertrophy — quantifies systolic and diastolic function, measures any left ventricular outflow tract gradient, looks for thrombus in LVNC, and identifies associated lesions. Serial echocardiography tracks progression and guides the timing of intervention. In a symptomatic child, the echo often confirms the diagnosis at the first encounter. [2] [9]
Cardiac magnetic resonance imaging adds tissue characterisation that echo cannot provide: late gadolinium enhancement maps the fibrosis that marks arrhythmic risk in HCM and ACM, and it clarifies morphologically borderline cases such as mild LVNC or apical HCM. Ambulatory Holter monitoring quantifies ventricular ectopy and non-sustained ventricular tachycardia, which feed the sudden-death risk stratification. Exercise testing documents functional capacity and the blood-pressure response (a blunted or falling blood pressure on exercise is an adverse sign in HCM). Cardiopulmonary exercise testing is used in older children to stage heart failure and to time transplant. [4] [5]
Genetic testing is now a core investigation, not a research add-on. A targeted cardiomyopathy gene panel identifies a pathogenic variant in roughly thirty to forty percent of affected children, with the highest yield in HCM. A positive result enables cascade screening of relatives, identifies genotype-positive phenotype-negative family members who need lifelong surveillance, and refines the prognosis. A negative result does not exclude a genetic cause. Genetic counselling before and after testing is essential because the result affects the whole family. Blood tests support the differential: viral studies and troponin for myocarditis, thyroid function, metabolic and lysosomal storage workup in infant or restrictive disease, and a full blood count and iron studies to exclude correctable contributors to failure. [4] [8]
Management — Resuscitation
The resuscitation scenario is the infant or child who presents in acute heart failure from a dilated cardiomyopathy. The priorities are to relieve congestion, improve perfusion, and support feeding and growth while the underlying diagnosis is confirmed and a definitive plan is built. Loop diuretics such as furosemide relieve pulmonary and systemic congestion; an angiotensin-converting-enzyme inhibitor reduces afterload; and, once the child is euvolaemic and stable, a carefully titrated beta-blocker and a mineralocorticoid antagonist are added as background heart-failure therapy, adapted from adult guidelines because high-quality paediatric evidence is limited. [7]
The paediatric evidence base for disease-modifying heart-failure drugs is sobering and must be known. Shaddy and colleagues' randomised controlled trial of carvedilol in children and adolescents with chronic heart failure — most of whom had DCM — found no significant benefit of carvedilol over placebo, in marked contrast to the adult trials. The trial does not prove carvedilol is useless in children, but it removed the confident claim of benefit and it is the answer an examiner expects when asked about paediatric heart-failure pharmacotherapy. The practical implication is that background therapy is continued, but the child is watched closely for non-response, because non-response in DCM is the signal to move toward transplant assessment. [7]
Furosemide (acute heart failure from DCM)
Loading dose
1 mg/kg PO/IV
Maintenance dose
1–2 mg/kg/day in 1–2 divided doses
Immediate management of the child presenting in heart failure
Assess ABC, perfusion, and feeding; weigh and plot growth centiles
Urgent ECG to exclude tachyarrhythmia (tachycardiomyopathy) and 12-lead for hypertrophy
Urgent echocardiography to confirm morphology, ejection fraction, and any obstruction
Relieve congestion with a loop diuretic; add an ACE inhibitor for afterload reduction
Once euvolaemic and stable, introduce beta-blocker and MRA with careful titration
Increase caloric density of feeds; consider nasogastric supplementation for FTT
Exclude mimics: viral myocarditis, thyroid disease, anaemia, metabolic disease
Refer to the paediatric heart-failure and transplant service; assess family
The resuscitation principle is that acute therapy is a bridge, not a destination. A child who does not improve, or who presents in fulminant failure, needs escalation to a tertiary centre with mechanical circulatory support capability, because the definitive treatment for end-stage DCM is transplantation, and a ventricular assist device may be needed as a bridge to it. [7]
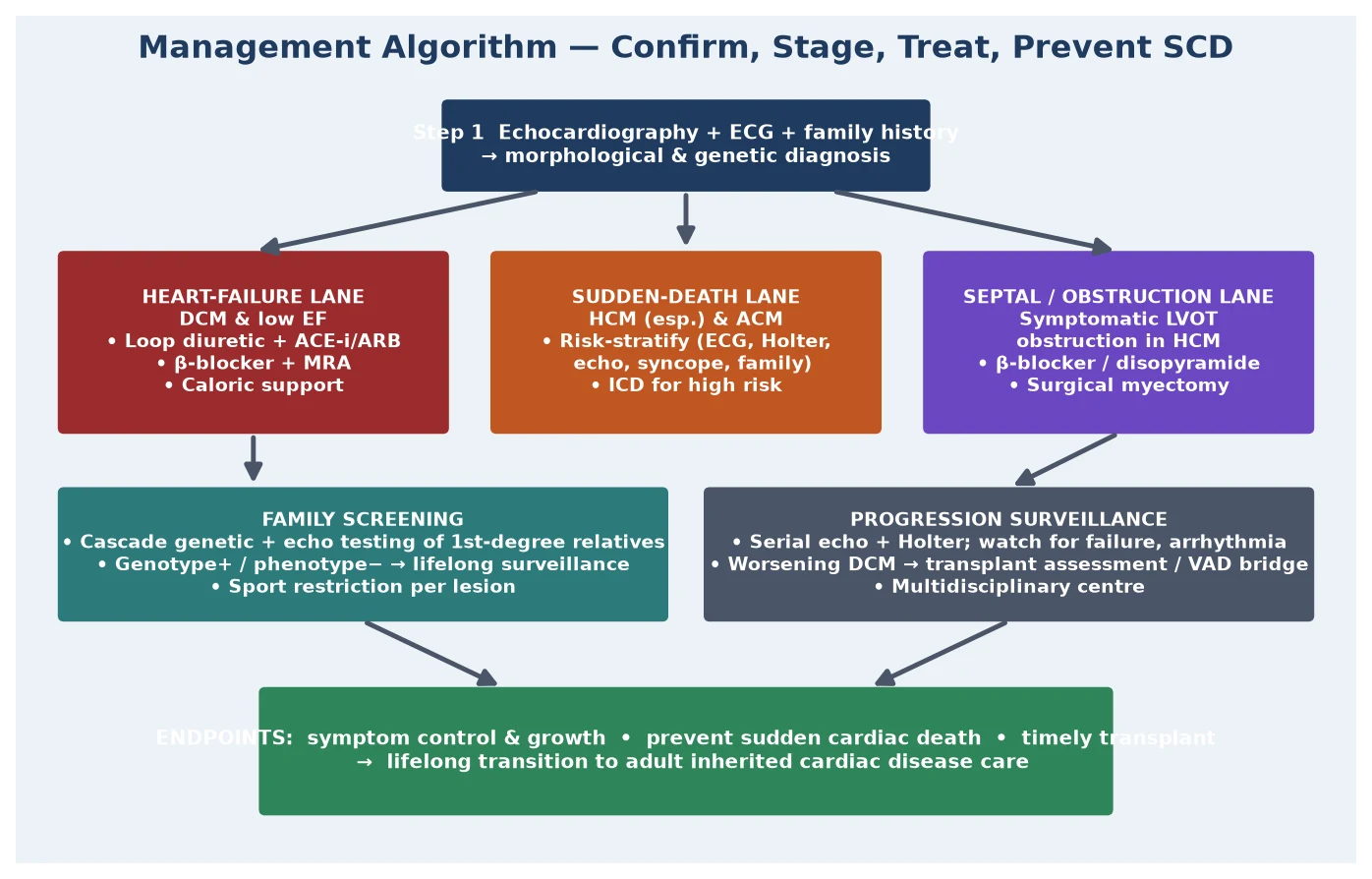
Management — Definitive & Stepwise
Definitive management is stratified by type and by the two outcomes the therapy is designed to prevent: progression to end-stage heart failure, and sudden cardiac death. The heart-failure lane governs DCM: diuretic, ACE inhibitor, beta-blocker, and MRA as background therapy; anticoagulation when the ejection fraction is severely depressed or there is atrial fibrillation; and, for the child who fails medical therapy, listing for cardiac transplantation, with a ventricular assist device as a bridge in the decompensating patient. DCM is the single largest indication for paediatric heart transplant. [7] [8]
The sudden-death lane governs HCM and ACM and is the most examined part of the topic. Risk stratification integrates the personal history (syncope), the family history (premature sudden death), the echocardiographic findings (massive hypertrophy, apical aneurysm), the Holter findings (non-sustained ventricular tachycardia), the exercise test (an abnormal blood-pressure response), and increasingly the cardiac MRI (extensive late gadolinium enhancement). Children who meet a high-risk threshold are offered an implantable cardioverter-defibrillator, the one intervention proven to reduce sudden death in HCM, on the strength of Maron and colleagues' landmark study. The device is not without cost — it carries a real risk of inappropriate shocks and lead complications in young, active patients — so the decision balances the absolute risk against the burden. [5] [9]
Symptomatic left ventricular outflow tract obstruction in HCM is managed in its own lane. The first-line drugs are beta-blockers and, if these are inadequate, disopyramide, both of which reduce the dynamic gradient by slowing the heart and increasing ventricular filling. Refractory obstructive symptoms in children are treated by surgical myectomy rather than the alcohol septal ablation used in adults, because the coronary anatomy in children makes ablation unsuitable. The rare infant with severe, symptomatic HCM from a RASopathy or metabolic disease is managed in a specialist centre, sometimes with specific therapy for the underlying syndrome. [4] [9]
[5] [7] [9]Specific Subtypes & Scenarios
Hypertrophic cardiomyopathy is the subtype an examiner most often expects in depth. It is usually autosomal dominant, caused by sarcomeric mutations in MYH7 or MYBPC3, and the long-term outcomes in childhood are now well described by Alexander and colleagues' national population-based study, which documented the cumulative incidence of death and transplant and identified the predictors of adverse outcome. The paediatric presentation differs from the adult: infants often present with heart failure rather than with sudden death, syndromic and metabolic causes are more common than in adults, and the phenotype can evolve as the child grows. The risk factors for sudden death in children overlap with the adult model but require paediatric-specific risk tools. [4] [9]
Dilated cardiomyopathy is the commonest type and the path to transplant. Burkett and Hershberger's review of familial DCM frames the genetic architecture — cytoskeletal, sarcomeric, and nuclear-envelope genes — and the practical point that a substantial fraction of apparently sporadic DCM is in fact familial once relatives are screened. The principal task in a new DCM is to exclude reversible mimics (tachycardiomyopathy, myocarditis, thyroid disease, anthracycline toxicity, metabolic disease), to start background therapy, to screen the family, and to identify the child who is heading toward transplant. [7] [8]
Restrictive cardiomyopathy, left ventricular non-compaction, and arrhythmogenic cardiomyopathy are individually rare but each carries a distinct lesson. Restrictive disease has the worst prognosis of all the cardiomyopathies, with high rates of sudden death and transplant, and it demands early transplant referral rather than prolonged medical therapy. Shi and colleagues' population study of childhood LVNC documented the long-term outcomes and the competing risks of death, transplant, and arrhythmia, and it guides surveillance and the decision to anticoagulate. Arrhythmogenic cardiomyopathy presents predominantly with arrhythmia and is managed with strict sport restriction and, for high-risk patients, an implantable defibrillator. [3] [10]
Across Australia, New Zealand, the United Kingdom and comparable high-resource systems, the model is a coordinated inherited cardiac conditions service. Every child with a cardiomyopathy is managed in a tertiary paediatric cardiology centre with access to cardiac MRI, genetic testing, electrophysiology, and heart-failure and transplant services. The Australian national population-based cohort studies (Alexander et al. for HCM, Shi et al. for LVNC) underpin the local outcome data and the surveillance intervals. Family cascade screening is organised centrally so that genotype-positive relatives are tracked, and transition to adult inherited cardiac disease care is planned in adolescence. Rural and remote children are supported by retrieval networks and telehealth links to the tertiary centre. [9] [10]
Complications & Pitfalls
The complications of a paediatric cardiomyopathy are the two endpoints the disease drives toward — progressive heart failure and sudden cardiac death — together with thromboembolism (especially in DCM with low ejection fraction and in LVNC), arrhythmia, infective endocarditis on the damaged endothelium, and the complications of treatment (device infections and inappropriate shocks from an implantable defibrillator, and the long-term burden of transplant including rejection, infection, and malignancy). The complications of restrictive disease deserve particular respect because both heart failure and sudden death can develop rapidly. [5] [10]
The pitfalls fall into diagnostic, family, and treatment errors. The diagnostic pitfall is to label a reversible tachycardiomyopathy as intrinsic DCM, or to attribute an athlete's symptoms to deconditioning and miss HCM. The family pitfall is to discharge relatives on a single normal echocardiogram without genetic testing or lifelong surveillance, because genotype-negative does not exclude disease and the phenotype can emerge with age. The treatment pitfall is twofold: to delay transplant assessment in a child who is failing medical therapy, and to place a defibrillator without weighing the burden of inappropriate shocks in a young patient. The overarching pitfall is to regard a cardiomyopathy as a static diagnosis rather than a dynamic disease that demands structured, lifelong surveillance. [5] [8]
Prognosis & Disposition
The prognosis is type-dependent and is the reason morphology matters so much at the bedside. Hypertrophic cardiomyopathy diagnosed in childhood has a substantial long-term burden of death and transplant, as documented by Alexander and colleagues' national population-based study, but the absolute annual rate of sudden death is low and concentrated in identifiable high-risk groups — which is why risk stratification and the defibrillator decision are the centre of management. Dilated cardiomyopathy prognosis is driven by the response to medical therapy: those who recover function do well, while non-responders progress to transplant, and DCM is the largest single indication for paediatric heart transplantation. [7] [9]
Restrictive cardiomyopathy has the worst prognosis of all, with high rates of both sudden death and transplant, and it justifies early transplant referral. Left ventricular non-compaction, characterised by Shi and colleagues' population outcomes, has a variable course that ranges from stable to progressive failure, arrhythmia, and transplant, with thromboembolism an additional threat. Across all types, the prognosis is improved by early diagnosis, family screening that catches at-risk relatives, rigorous risk stratification for sudden death, and timely transplant for end-stage disease. [3] [10]
Disposition is always to a tertiary paediatric cardiology centre with inherited cardiac conditions expertise. The confirmed case is enrolled in structured surveillance with serial echocardiography, ECG, and Holter monitoring; the high-risk patient has a defibrillator and sport restriction; the failing patient is listed for transplant; and every family undergoes cascade genetic and clinical screening. Transition to adult inherited cardiac disease care is planned in adolescence, because the disease and its surveillance are lifelong. [9] [10]
Special Populations
The genotype-positive phenotype-negative relative is the first special population and the most important to manage correctly. A first-degree relative who carries the family's pathogenic variant but has a normal echocardiogram is not "reassured and discharged" — they are placed on structured lifelong surveillance, because the phenotype can emerge at any age. The surveillance interval is set by the gene, the age, and the family phenotype. Conversely, a genotype-negative first-degree relative of a genotype-positive proband can usually be released from surveillance, which is one of the great practical benefits of genetic testing. [4] [8]
The infant with metabolic or syndromic cardiomyopathy is the second. Inborn errors of metabolism, mitochondrial disease, Barth syndrome, and the RASopathies (Noonan and related) each produce distinctive cardiomyopathy phenotypes and carry extracardic implications that shape management — immunology, growth, developmental, and anaesthetic considerations. These infants are managed jointly with metabolic medicine and clinical genetics, because some underlying disorders are partially treatable and the prognosis is set as much by the systemic disease as by the heart. [4] [9]
The competitive young athlete with possible cardiomyopathy is the third and the most publicly visible. The central question is whether an apparent athlete's heart masks HCM, because competitive sport accelerates the disease and raises the sudden-death risk. The evaluation rests on the ECG, the echo, the family history, and genetic testing, and the default when uncertainty cannot be resolved is restriction from competitive sport, which is a decision with profound personal consequences and must be made in a specialist inherited cardiac conditions service. The rural or remote child with cardiomyopathy is supported by retrieval networks and telehealth, ensuring the same standard of surveillance as a city child. [5] [6]
Evidence, Guidelines & Regional Differences
The evidence base rests on incidence, classification, sudden death, treatment, and outcomes. The incidence reference is the Lipshultz Pediatric Cardiomyopathy Registry study, which established the ~1 per 100 000 per year figure. The classification references are the 2006 American Heart Association scientific statement of Maron and colleagues and the 2008 European Society of Cardiology position statement of Elliott and colleagues, which together define the morphological framework used worldwide. [1] [2] [3]
The sudden-death evidence comes from Maron's systematic review of HCM, Maron and colleagues' randomised evidence for the implantable defibrillator in high-risk HCM, and Bagnall and colleagues' prospective study of sudden cardiac death in children and young adults, which established the contribution of inherited cardiac conditions — cardiomyopathy foremost — to sudden death in the young. The treatment evidence is anchored by Burkett and Hershberger's review of familial DCM and, crucially, by the Shaddy carvedilol trial, the randomised paediatric heart-failure study that found no significant benefit and tempered the translation of adult heart-failure pharmacotherapy to children. [4] [5] [6] [7] [8]
The outcomes evidence is the strength of the ANZ literature: the national population-based studies of Alexander and colleagues (childhood HCM) and Shi and colleagues (childhood LVNC) give local, unbiased outcome data that underpin surveillance intervals and transplant timing. The guideline landscape is shared between the American Heart Association and the European Society of Cardiology, with the Australian and New Zealand inherited cardiac conditions services operating to the same principles. The central consensus across regions is uniform: classify morphologically, test genetically, screen the family, risk-stratify for sudden death, treat heart failure, and transplant for end-stage disease. [9] [10]
Across ANZ, the United Kingdom, Europe and North America, the consensus model is the same in principle: a coordinated inherited cardiac conditions service that integrates paediatric cardiology, clinical genetics, cardiac MRI, electrophysiology, and heart-failure and transplant services, with central organisation of family cascade screening and planned transition to adult care. The thresholds for defibrillator implantation, the surveillance intervals, and the transplant timing vary in detail by guideline body and centre, but the strategy — morphology first, genotype second, screen the family, prevent sudden death, transplant for end-stage — is uniform across high-resource settings. [2] [9] [10]
Exam Pearls
Cardiomyopathy types — 'D-H-R-N-A' morphology
References
- [1]Lipshultz SE; Sleeper LA; Towbin JA; Lowe AM; Orav EJ; Cox GF; et al The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med, 2003.PMID 12711739
- [2]Maron BJ; Towbin JA; Thiene G; Antzelevitch C; Corrado D; Arnett D; et al Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement. Circulation, 2006.PMID 16567565
- [3]Elliott P; Andersson B; Arbustini E; Bilinska Z; Cecchi F; Charron P; et al Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J, 2008.PMID 17916581
- [4]Maron BJ Hypertrophic cardiomyopathy: a systematic review. JAMA, 2002.PMID 11886323
- [5]Maron BJ; Shen WK; Link MS; Epstein AE; Almquist AK; Daubert JP; et al Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med, 2000.PMID 10666426
- [6]Bagnall RD; Weintraub RG; Ingles J; Duflou J; Yeates L; Lam L; et al A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N Engl J Med, 2016.PMID 27332903
- [7]Shaddy RE; Boucek MM; Hsu DT; Boucek RJ; Canter CE; Mahony L; et al Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA, 2007.PMID 17848651
- [8]Burkett EL; Hershberger RE Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol, 2005.PMID 15808750
- [9]Alexander PMA; Nugent AW; Daubeney PEF; Lee KJ; Sleeper LA; Schuster T; et al Long-Term Outcomes of Hypertrophic Cardiomyopathy Diagnosed During Childhood: Results From a National Population-Based Study. Circulation, 2018.PMID 29490994
- [10]Shi WY; Moreno-Betancur M; Nugent AW; Cheung M; Colan S; Turner C; et al Long-Term Outcomes of Childhood Left Ventricular Noncompaction Cardiomyopathy: Results From a National Population-Based Study. Circulation, 2018.PMID 29514799