Paeds · cardiology
Coarctation and interrupted aortic arch
Also known as Coarctation of the aorta · Interrupted aortic arch · IAA · Ductal-dependent arch obstruction · Aortic isthmus stenosis · Hypoplastic aortic arch
Fellowship guide to coarctation of the aorta and interrupted aortic arch: the two ductal-dependent left-heart-outflow obstructions that declare themselves as neonatal collapse when the duct closes, the four-limb blood-pressure gradient and weak femoral pulses at the bedside, prostaglandin E1 as the non-negotiable bridge to surgery, and the syndromic associations (Turner, 22q11.2, bicuspid aortic valve) that every candidate must name.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single idea that organises the whole topic is that coarctation and IAA are the same disease at different severities: both obstruct the aortic arch, both depend on the duct for descending-aortic flow, and both declare themselves at ductal closure. Coarctation narrows the isthmus; IAA interrupts it. The clinical picture, the resuscitation and the surgical logic flow from that one mechanism. [1] [6]
This page covers the recognition and neonatal management of both lesions, the syndromic associations that change the work-up (Turner syndrome, 22q11.2 deletion, bicuspid aortic valve), the surgical options and their complications, and the lifelong follow-up that every repaired patient needs. It links to the ductal-dependent congenital heart disease and neonatal cyanosis leaves for the broader differential. [2] [5]
Overview & Definition
Coarctation of the aorta is a discrete shelf-like infolding of the media at the aortic isthmus — the segment between the left subclavian artery and the ductus arteriosus — that narrows the lumen and obstructs forward flow to the descending aorta. Interrupted aortic arch is the extreme of the same spectrum: the arch is completely discontinuous, so the descending aorta receives blood only through the ductus arteriosus. [1] [2]
What sets these lesions apart from other left-heart obstructions is their ductal dependence. In fetal life the duct carries right ventricular output to the descending aorta, bypassing the narrowed or interrupted isthmus entirely, so the fetus grows normally and the lesion is invisible at birth. The disease declares itself only when the duct constricts over the first hours to days of life — suddenly cutting off the only route to the descending aorta and causing acute inferior-body hypoperfusion. This is why the classic neonatal coarctation or IAA presentation is a well baby who collapses on day three or four. [3] [4]
The two lesions sit within the broader family of ductal-dependent congenital heart disease, alongside hypoplastic left heart syndrome, critical aortic stenosis and transposition. The unifying principle is that the neonate is stable only while the duct is open, so prostaglandin E1 is the first drug in any neonate who collapses after a period of wellbeing and shows shock, differential cyanosis or weak pulses. [4] [7]
Classification
Sort the disease by the anatomy of the obstruction, because the anatomy sets both the timing of presentation and the surgical approach. The figure below splits arch obstruction into coarctation (a narrowing classified by its site relative to the ductus) and interrupted aortic arch (a gap classified by the Celoria and Patton level of interruption). [1] [6]
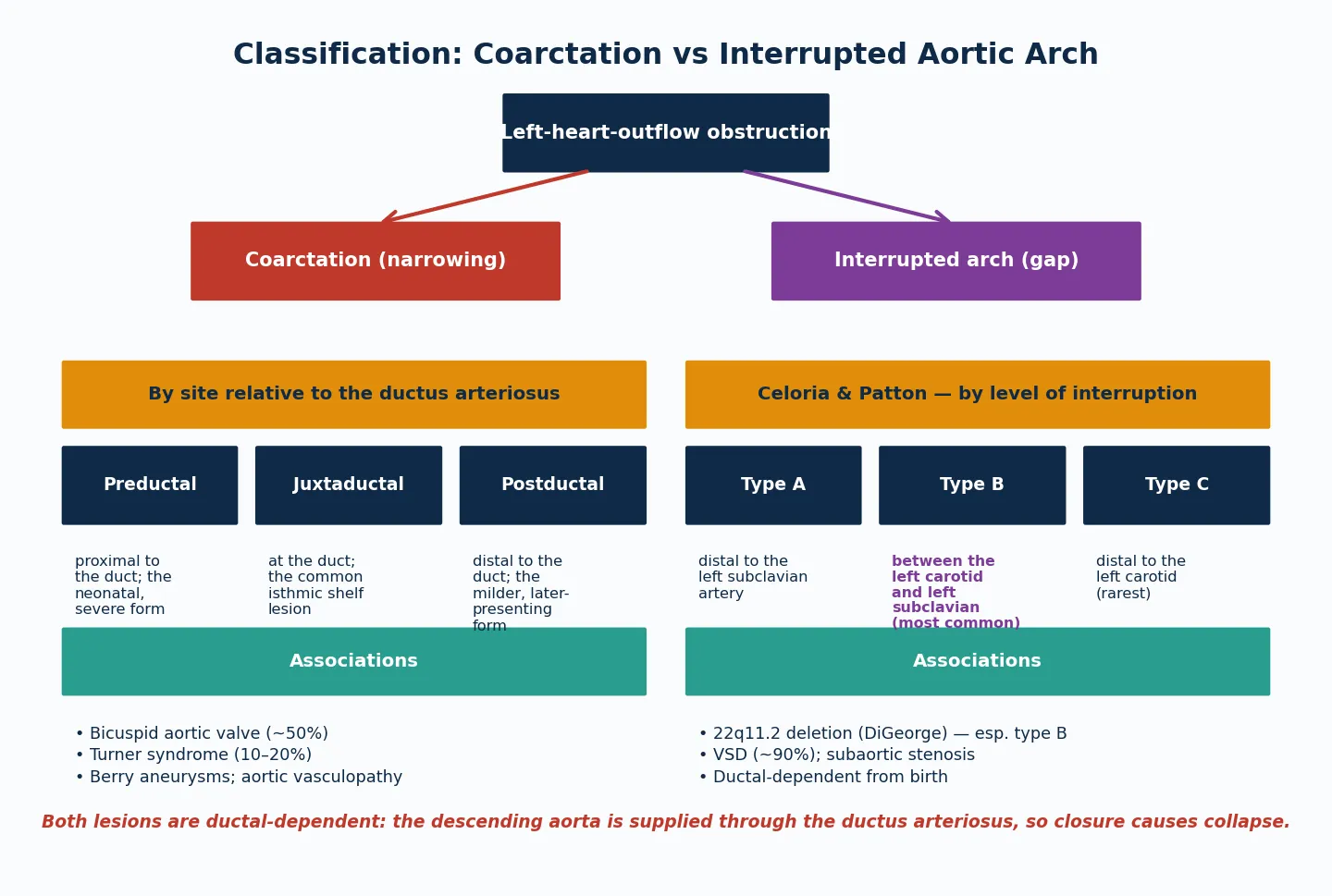
Coarctation (narrowing)
- Discrete shelf at the isthmus; juxtaductal is the common form
- Bicuspid aortic valve in ~50% — examine and image the valve
- Turner syndrome in 10–20% — request karyotype in any girl
- Presents neonatally (severe) or later in childhood (milder)
Interrupted aortic arch (gap)
- Complete discontinuity; type B (distal to left subclavian origin, between carotid and subclavian) is most common
- VSD in ~90%; subaortic stenosis frequent
- 22q11.2 deletion strongly associated, especially type B
- Always ductal-dependent from birth — no late presentation
Shared features
- Both supplied through the ductus arteriosus in fetal life
- Both declare at ductal closure: shock, acidosis, weak femorals
- Both respond to prostaglandin E1 — the bridge to surgery
- Both need lifelong follow-up for re-obstruction and hypertension
The Celoria and Patton classification of IAA names the level at which the arch is interrupted. Type A sits distal to the left subclavian artery; type B sits between the left carotid and the left subclavian and is the most common; type C sits distal to the left carotid and is the rarest. Type B carries the strongest association with 22q11.2 deletion, which is why a chromosomal microarray is part of the first work-up in any IAA diagnosis. [5] [8]
Epidemiology & Risk Factors
Coarctation of the aorta accounts for roughly five to eight percent of all congenital heart defects, making it one of the commoner lesions a general paediatrician will encounter. Interrupted aortic arch is far rarer, representing under one percent of congenital heart disease, but its severity and ductal dependence make it disproportionately important in neonatal cardiology. [1] [2]
The dominant associations that a fellowship candidate must name are bicuspid aortic valve, Turner syndrome and 22q11.2 deletion. About half of coarctation patients have a bicuspid aortic valve, which itself drives aortic vasculopathy and later valve disease. Turner syndrome co-occurs with coarctation in a substantial fraction of girls, so any female presenting with coarctation earns a karyotype. 22q11.2 deletion is the signature genetic association of interrupted aortic arch, particularly type B. [9] [12]
The reason these syndromic associations matter clinically is that they change the management beyond the heart. Turner syndrome carries gonadal and growth implications that need endocrine input. 22q11.2 deletion carries hypocalcaemia, immunodeficiency and airway anomalies that change the anaesthetic and intensive-care plan. Failing to recognise the syndrome means failing to prepare for complications that are entirely preventable. [8] [9]
Pathophysiology
To see why a coarctation or IAA baby collapses, picture the fetal circulation as a system that routes blood around the obstruction through the ductus arteriosus. In utero, the right ventricle pumps through the pulmonary artery and across the duct into the descending aorta, bypassing the narrowed isthmus. The fetus thrives because the duct does the work of the isthmus. [3] [4]
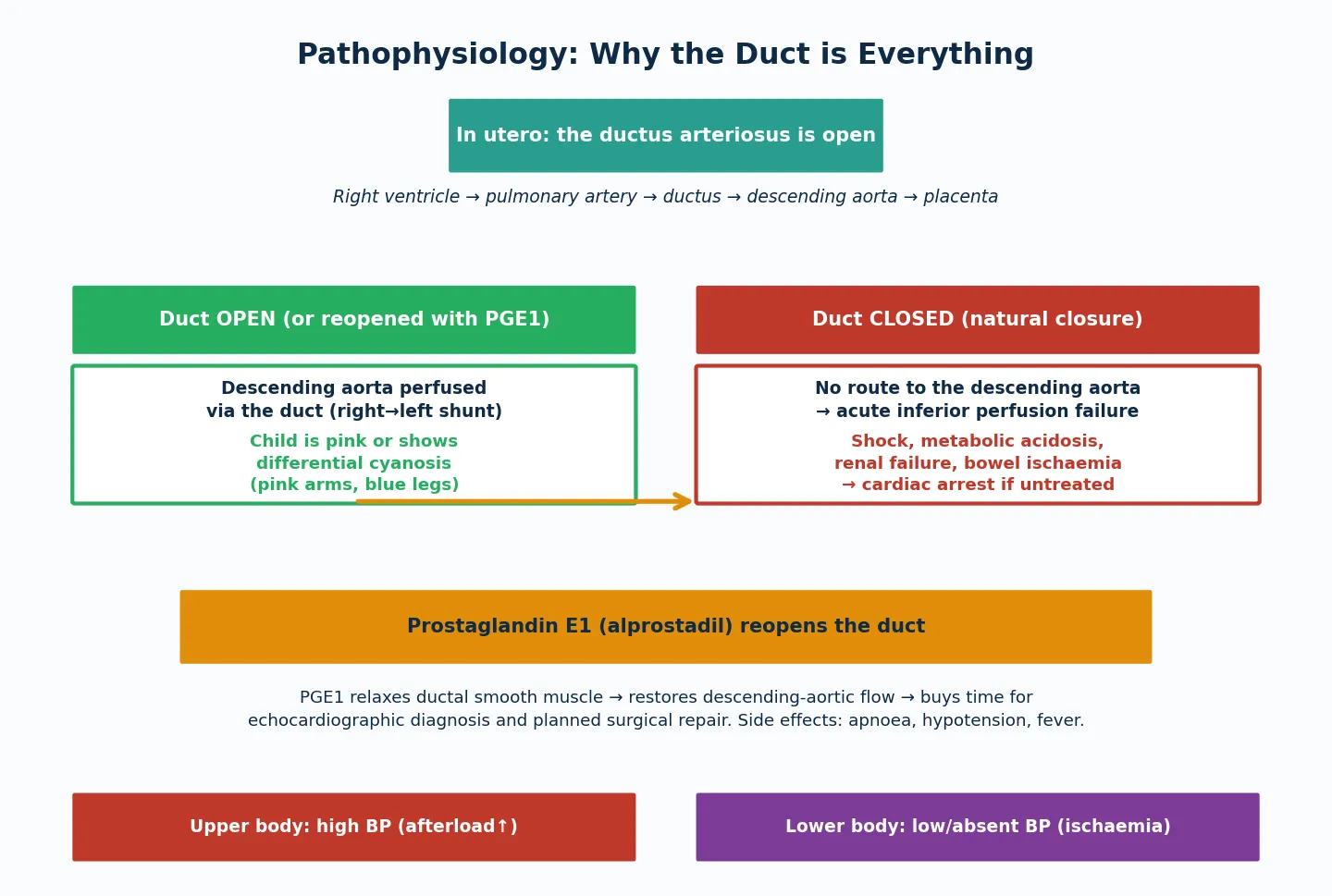
The collapse happens because ductal constriction is a biological clock, not a random event. As oxygen tension rises after birth, the ductal smooth muscle constricts over hours to days, progressively narrowing the shunt. In a normal baby this is harmless, but in a coarctation or IAA baby it progressively chokes the only supply to the descending aorta. The child develops progressive acidosis, oliguria from renal hypoperfusion, and poor perfusion, then cardiovascular collapse. The window between the first signs and cardiac arrest can be short. [4] [7]
Prostaglandin E1 (alprostadil) works by relaxing ductal smooth muscle to reopen the duct, restoring descending-aortic flow and reversing the shock. It is the single most important drug in neonatal ductal-dependent heart disease, and its mechanism — direct relaxation of the ductal wall — explains why it works within minutes and why its side effects (apnoea, hypotension, fever) are predictable and manageable. The practical rule is to start it empirically in any collapsing neonate with a ductal-dependent picture, before the echocardiogram confirms the diagnosis. [4] [3]
Clinical Presentation
A neonate with coarctation or IAA usually presents in one of two ways: the dramatic neonatal collapse, or the subtle bedside finding that an alert clinician catches before collapse. The dramatic presentation is a baby who was feeding and thriving for the first two to four days of life, then becomes tachypnoeic, mottled and oliguric, progressing to shock and metabolic acidosis as the duct constricts. This is the classic day-three-to-four collapse, and the single most important bedside sign is weak or absent femoral pulses in a shocked neonate. [3] [4]
The subtler presentation is the older infant or child found to have hypertension on a routine check, or referred for a murmur or failure to thrive. The femoral pulses are weak or delayed relative to the radial (radio-femoral delay), and four-limb blood pressures show an arm-to-leg systolic gradient. A systolic murmur may be audible at the left sternal border and over the back, and prominent interscapular pulsations may be felt where collateral vessels develop around the obstruction. [1] [2]
Differential cyanosis — a pink upper body with a blue lower body — is the pathognomonic finding when the duct is still open but the obstruction is severe. It arises because well-oxygenated blood from the left ventricle reaches the head and upper limbs across the proximal arch, while deoxygenated right ventricular blood reaches the lower body through the duct. Checking pre-ductal (right arm) and post-ductal (leg) saturations side by side reveals the gradient and should be routine in any neonate with suspected cardiac disease. [2] [3]
Differential Diagnosis
The differential turns on separating ductal-dependent arch obstruction from the other causes of neonatal shock, and from the other causes of hypertension in an older child. The key distinction at the bedside is the pulse pattern: weak femoral pulses with good upper-body pulses point to arch obstruction, whereas uniformly weak pulses point to septic shock or a myocarditis. [2] [3]
Points to arch obstruction
- Weak or absent femoral pulses with strong brachial/radial pulses
- Four-limb BP: arm-to-leg systolic gradient above 20 mmHg
- Differential cyanosis (pink arms, blue legs) when duct open
- Well for first days then collapse at ductal closure
- Abnormal four-chamber or arch view on antenatal scan
Points to a mimic
- Septic shock: uniformly weak pulses, fever, normal saturations
- Hypoplastic left heart: grey collapse, poor all pulses, mitral/aortic atresia
- Critical aortic stenosis: murmur, pulmonary oedema, weak all pulses
- Essential or renal hypertension (older child): normal pulses, no gradient
- Cardiomyopathy or myocarditis: gallop, hepatomegaly, globally poor function
In the older child or adolescent with hypertension, the critical step is always to measure four-limb blood pressures and palpate the femoral pulses. An arm-to-leg gradient or radio-femoral delay makes coarctation the working diagnosis until echocardiography excludes it. Failing to measure leg pressures in a hypertensive child is the classic missed-diagnosis error, because coarctation is a surgically curable cause of hypertension. [1] [11]
Antenatal diagnosis increasingly shifts the presentation from collapse to planned management. When a suspected coarctation or IAA is identified on the fetal anomaly or cardiac scan, the baby is delivered at a cardiac centre, prostaglandin E1 is started prophylactically or at the first sign, and the echocardiogram confirms the anatomy before any deterioration. Prenatal suspicion does not eliminate neonatal collapse entirely, but it shortens the time to definitive treatment. [3] [8]
Clinical & Bedside Assessment
The focused assessment of a neonate or child with suspected arch obstruction rests on three bedside manoeuvres: palpate all four limb pulses, measure four-limb blood pressures, and check pre- and post-ductal saturations. In a shocked neonate, weak or absent femoral pulses with bounding upper-limb pulses is the arch-obstruction signature, and it should trigger prostaglandin E1 before any further investigation. [2] [4]
Measure four-limb blood pressures with appropriately sized cuffs. An arm-to-leg systolic gradient above twenty millimetres of mercury, with the arm pressure higher, supports coarctation. Note that in a shocked neonate with poor perfusion all pressures may be low and the gradient may be masked, so do not be falsely reassured by an unremarkable gradient in a collapsed baby — trust the pulse pattern and the clinical picture. [1] [11]
Examine for the syndromic stigmata that flag the associated conditions. Short stature, webbed neck, widely spaced nipples and a low posterior hairline suggest Turner syndrome in any girl with coarctation. Cleft palate, a small chin, low-set ears and hypocalcaemia suggest 22q11.2 deletion in an infant with IAA. Listen for the systolic murmur of a bicuspid aortic valve or a VSD, and feel for the heaving apex of left ventricular pressure overload. A thorough cardiac and dysmorphology examination at the bedside often predicts the echocardiographic findings. [9] [8]
Investigations
Echocardiography is the definitive investigation and usually the only imaging needed before surgery. A complete study defines the site and severity of the obstruction, the arch anatomy (including the distance between the head and neck vessels in IAA), the presence and size of a ventricular septal defect, the aortic valve morphology (bicuspid or not), the ductal patency, and the direction of ductal flow. It also assesses ventricular function and excludes associated lesions. [2] [5]
Echocardiography
- First-line and usually definitive — defines the whole anatomy
- Shows site and severity of narrowing or interruption
- Assesses arch, VSD, aortic valve, duct, ventricular function
- Doppler estimates the gradient across the obstruction
Supporting bloods
- Blood gas: metabolic acidosis with raised lactate in collapse
- Renal function: acute kidney injury from hypoperfusion
- Calcium: check for hypocalcaemia of 22q11.2 deletion
- CBC and CRP: exclude sepsis as a differential
Genetic and adjunct tests
- Karyotype in any girl with coarctation (Turner syndrome)
- Chromosomal microarray or FISH for 22q11.2 in IAA
- Chest X-ray: rib notching in older children (collaterals)
- MRI or CT angiogram for complex or post-operative anatomy
Blood gas and biochemistry assess the severity of the insult and guide resuscitation. A metabolic acidosis with a raised lactate reflects the degree of tissue hypoperfusion and should improve as prostaglandin E1 restores descending-aortic flow. Renal function may show acute kidney injury from hypoperfusion, and the calcium should be checked in any infant with IAA because 22q11.2 deletion causes hypocalcaemia that can complicate the perioperative course. [7] [8]
Genetic testing is part of the first work-up, not an add-on. In a girl with coarctation, a karyotype screens for Turner syndrome, because the diagnosis drives endocrine and reproductive follow-up. In any infant with interrupted aortic arch, chromosomal microarray or fluorescence in-situ hybridisation screens for 22q11.2 deletion, because the deletion changes the surgical, airway, calcium and immunology management. Chest X-ray in the older child may show rib notching from collateral vessels eroding the undersurface of the ribs, a classic but late sign. [9] [8]
Management — Resuscitation
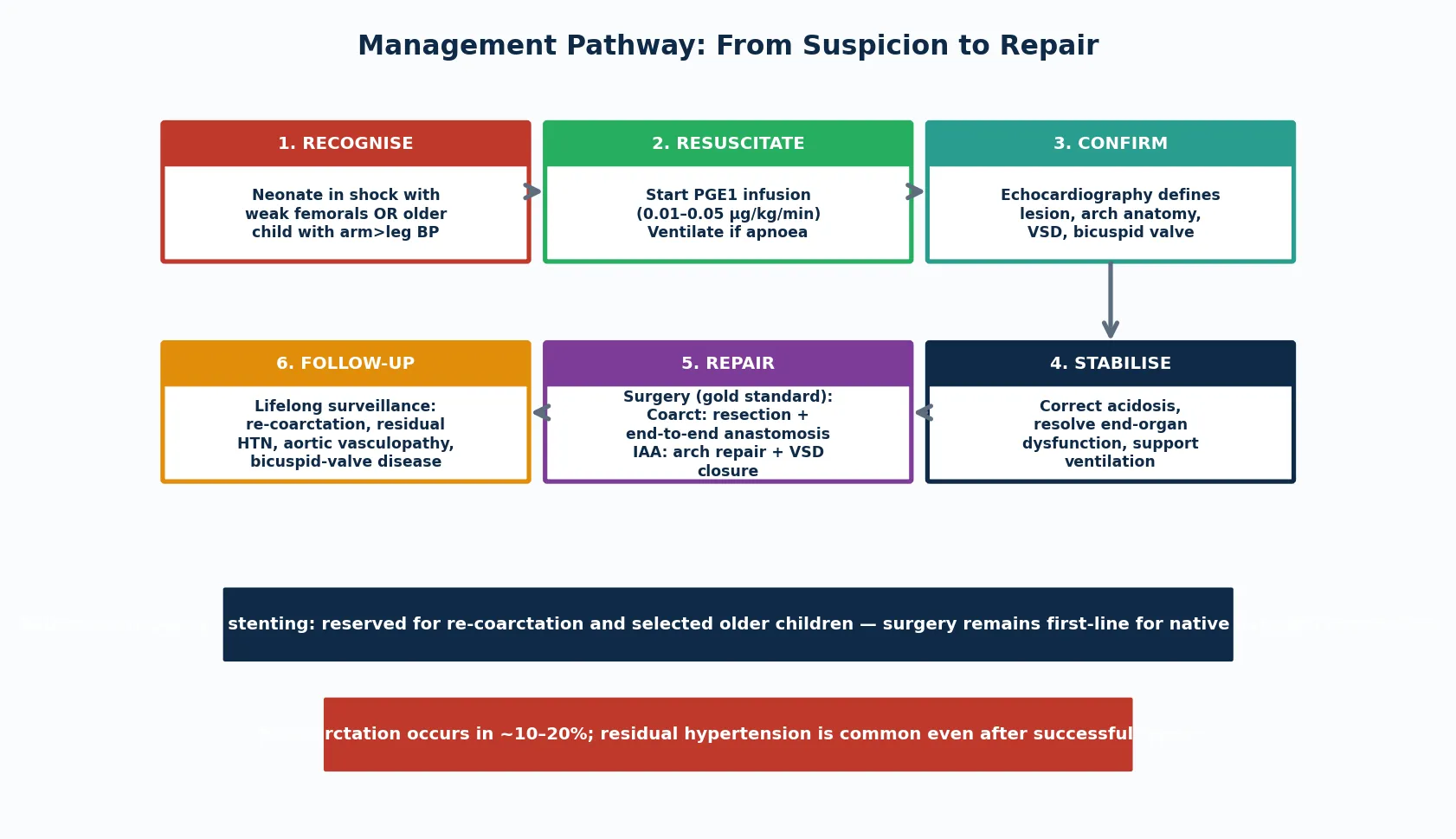
The resuscitation of a neonate with ductal-dependent arch obstruction rests on one drug and one principle: prostaglandin E1 to reopen the duct, started before the diagnosis is confirmed. A collapsing neonate with weak femoral pulses gets the infusion empirically — the downside of an unnecessary dose (apnoea, hypotension) is manageable, while the downside of delay is death. [4] [3]
Prostaglandin E1 (alprostadil) is started at 0.01 to 0.05 micrograms per kilogram per minute and titrated to the clinical response. The duct usually reopens within thirty minutes to a few hours, restoring descending-aortic flow, improving perfusion and correcting the metabolic acidosis. Apnoea is the most important side effect: roughly one in ten infants on PGE1 needs intubation, so the team must be ready to ventilate, and a baby on PGE1 being retrieved to a cardiac centre must travel intubated or with skilled airway support. [4] [7]
Alongside PGE1, correct the metabolic acidosis with fluid and inotropic support as needed, and treat the renal and electrolyte consequences of the hypoperfusion. Avoid giving large volumes of fluid blindly, because the left ventricle may be pressure-loaded and volume-sensitive; guide resuscitation by clinical perfusion, lactate trend and, once available, central venous and arterial pressures. Once the child is stabilised on PGE1, the echocardiogram defines the anatomy and the surgical plan follows. [3] [7]
Management — Definitive & Stepwise
Definitive treatment is surgical repair, and the approach depends on the lesion and the anatomy. For coarctation, the gold standard in neonates and infants is resection of the narrowed segment with end-to-end anastomosis, which removes the ductal tissue entirely and reduces the risk of re-coarctation. Extended end-to-end and arch advancement techniques are used when the arch is hypoplastic. The surgery is done through a left thoracotomy without cardiopulmonary bypass. [2] [6]
For interrupted aortic arch, the repair is more complex because the arch must be reconstructed and the almost invariable VSD closed. The modern approach is single-stage complete repair — restoring arch continuity (usually by direct anastomosis of the descending to the ascending aorta) and closing the VSD in one operation — though some centres use a staged approach. The surgery requires cardiopulmonary bypass and circulatory arrest, and the presence of 22q11.2 deletion, subaortic stenosis and airway anomalies adds perioperative complexity. [5] [6]
Balloon angioplasty and stenting have a defined but secondary role. For native (previously untreated) coarctation in neonates and infants, surgery remains first-line because balloon angioplasty carries a higher recurrence rate in this age group. Balloon angioplasty and stenting are reserved for re-coarctation after surgical repair and for selected older children and adolescents, where they avoid a repeat thoracotomy and have good medium-term results. The systematic-review evidence supports this surgical-first approach for native neonatal coarctation. [10] [2]
Specific Subtypes & Scenarios
Neonatal coarctation presenting as collapse is the scenario the acute exam tests. A baby well for two to four days then presents in shock with metabolic acidosis and weak femoral pulses. The key is to recognise the ductal-dependent picture, start prostaglandin E1 empirically, and arrange echocardiographic confirmation and transfer to a cardiac centre. The pitfall is attributing the collapse to sepsis alone and missing the weak femoral pulses, which delays the PGE1 that would save the child. [3] [4]
Turner syndrome and coarctation is the scenario the short case tests. A girl with short stature, webbed neck and coarctation has Turner syndrome until the karyotype proves otherwise, because the cardiovascular risk (coarctation, bicuspid valve, aortic dissection) and the endocrine needs (growth hormone, gonadal dysgenesis) both depend on the diagnosis. Every girl with coarctation earns a karyotype, and the echocardiogram should specifically assess the valve and the aortic root, because aortic vasculopathy increases the lifetime dissection risk. [9] [12]
Interrupted aortic arch type B with 22q11.2 deletion is the scenario that tests the genetics link. Type B IAA (interruption between the left carotid and left subclavian) carries the strongest association with 22q11.2 deletion, so chromosomal microarray is part of the first work-up. The deletion brings hypocalcaemia (needing calcium monitoring and replacement), immunodeficiency (affecting transfusion and vaccination), palatal and airway anomalies (affecting anaesthesia) and neurodevelopmental risk, all of which change the perioperative and long-term plan. Prenatal diagnosis and early repair have improved outcomes substantially. [8] [7]
The older child or adolescent with residual hypertension after repair is the follow-up scenario. Even after a successful coarctation repair, a substantial proportion of patients have residual or recurrent hypertension, driven by an intrinsic aortic vasculopathy rather than residual obstruction alone. This is why lifelong blood-pressure surveillance is non-negotiable, and why repaired coarctation patients are not discharged from cardiac follow-up at any age. [1] [11]
Complications & Pitfalls
The untreated arch obstruction carries a heavy burden in the neonate: hypoxic-ischaemic injury to the brain, kidneys and gut from the low-output state; acute kidney injury; and death from cardiovascular collapse. Many of these are preventable with timely PGE1, which is why the single biggest modifiable prognostic factor is the speed of recognition and ductal reopening. [4] [3]
After repair, the dominant complications are re-coarctation and residual hypertension. Re-coarctation occurs in roughly ten to twenty percent of surgically repaired patients, more often in those repaired as small neonates, and is treated with balloon angioplasty or stenting. Residual hypertension is common even after a haemodynamically successful repair and reflects the intrinsic aortic vasculopathy, which is why lifelong blood-pressure monitoring is mandatory. [2] [11]
The treatment carries its own pitfalls. PGE1 apnoea in transit is a recognised preventable death, so any baby on PGE1 being retrieved must travel intubated or with airway expertise. Missing the karyotype in a girl with coarctation denies her the Turner-syndrome surveillance that prevents aortic dissection and addresses growth and gonadal needs. Failing to test for 22q11.2 in an IAA baby leaves hypocalcaemia, immunodeficiency and airway anomalies unanticipated in theatre. Each is an avoidable harm rooted in forgetting the disease's associations. [9] [8]
Prognosis & Disposition
With timely prostaglandin stabilisation and expert surgical repair, the early survival of both coarctation and interrupted aortic arch is excellent in the modern era. Prenatal diagnosis, improved surgical techniques and better perioperative care have pushed operative mortality for isolated coarctation to very low levels, and even the more complex IAA repair now carries good short-term survival when performed in a specialist centre. [6] [8]
The long-term battleground is the late complications. Re-coarctation, residual and recurrent hypertension, aortic vasculopathy with dissection risk, and bicuspid aortic valve disease (with its own stenosis and regurgitation trajectory) all declare over decades, not days. Long-term follow-up series show that a significant proportion of repaired coarctation patients have hypertension by young adulthood, and that premature cardiovascular disease is the dominant late concern. This is why repaired patients are never discharged from cardiac surveillance. [1] [11]
Disposition is to a tertiary paediatric cardiac centre for the acute repair, then to a structured transition pathway into adult congenital heart disease services. The general paediatrician's role is to recognise the lesion early, start PGE1, coordinate the retrieval, and then champion the lifelong follow-up that the patient needs across the transition to adult care. [2] [12]
Special Populations
Neonates with prenatal diagnosis increasingly present as a managed rather than an emergency cohort. When a suspected arch obstruction is identified antenatally, the baby is delivered at or near a cardiac centre, the team is prepared, and PGE1 is started at the first clinical sign. This shifts the outcome by eliminating the delay between collapse and treatment, though even prenatally suspected cases need vigilant neonatal assessment because the diagnosis is not always confirmed. [3] [8]
Girls with Turner syndrome and coarctation carry the combined cardiovascular burden of arch obstruction, bicuspid valve, and aortic vasculopathy, alongside the endocrine and reproductive needs of the syndrome. They need coordinated cardiology, endocrinology (growth hormone, oestrogen replacement) and reproductive input, and lifelong aortic surveillance because the dissection risk persists after repair. [9] [12]
Infants with 22q11.2 deletion and IAA face the cardiac surgery alongside the multisystem burden of the deletion — hypocalcaemia, immunodeficiency, palatal and airway anomalies, feeding difficulty and neurodevelopmental risk. The perioperative and long-term management must integrate cardiology, immunology, endocrinology, speech and language therapy, and developmental paediatrics. Remote and Indigenous families may face the logistics of prolonged separation from home and community during the surgical admission, so a clear retrieval and repatriation pathway, telehealth follow-up and cultural support are essential. [8] [7]
Evidence, Guidelines & Regional Differences
The evidence base for coarctation spans modern lifespan reviews and long-term follow-up cohorts. The lifespan review by Salciccioli and Zachariah (2023) sets out the contemporary paradigms for coarctation across the age range, and the clinical review by Kim and colleagues (2020) covers the cardiology-clinic perspective. The long-term follow-up study of Toro-Salazar and colleagues (2002) anchors the late-hypertension and cardiovascular-risk data that justify lifelong surveillance. [1] [2]
For interrupted aortic arch, the surgical overview of Schreiber and colleagues (1997) remains a foundational reference for the classification and surgical evolution, while the surgical-considerations review of LaPar and Baird (2018) and the perioperative and anaesthetic review of Burbano-Vera and colleagues (2018) cover the modern operative and intensive-care approach. The 22q11.2-deletion cohort study of Ron and colleagues (2022) documents the improved outcomes from prenatal and early postnatal diagnosis. The Turner-syndrome study of Eckhauser and colleagues (2015) anchors the genetics link for coarctation. [5] [6]
Regional practice differences are modest because the surgical and resuscitation principles are internationally adopted, but access to paediatric cardiac surgery, neonatal retrieval and PGE1 in remote areas varies. In Australia and New Zealand, suspected ductal-dependent arch obstruction is referred to a tertiary paediatric cardiac centre, with retrieval services trained to start PGE1 in transit and intubate prophylactically. The main controversies are the timing and type of repair (single-stage versus staged for IAA), the role of primary balloon angioplasty in native coarctation, and the optimal surveillance intensity for late hypertension and aortic vasculopathy. [2] [10]
Exam Pearls
Hold one sentence for the viva: a neonate who was well then collapses on day three or four with weak femoral pulses has a ductal-dependent arch obstruction until proven otherwise, and the treatment is prostaglandin E1 started before the echocardiogram confirms the diagnosis. [4] [3]
State the frequently tested facts correctly. The classic age of collapse is day three to four, at ductal closure. The bedside signs are weak or absent femoral pulses and a four-limb blood-pressure arm-to-leg gradient. Prostaglandin E1 at 0.01 to 0.05 micrograms per kilogram per minute reopens the duct. Bicuspid aortic valve coexists in about half. Turner syndrome earns a karyotype in any girl with coarctation. 22q11.2 deletion is the genetic signature of IAA, especially type B. Surgery is first-line for native neonatal coarctation; balloon angioplasty is for re-coarctation. [1] [9]
The high-yield pairings: a girl with coarctation is Turner syndrome until karyotyped; an infant with type B IAA is 22q11.2 deletion until tested; an older child with hypertension and radio-femoral delay is coarctation until echo excludes it; a repaired patient with high blood pressure has intrinsic aortic vasculopathy, not necessarily re-obstruction. These pairings do most of the diagnostic work in the short and long cases. [11] [12]
References
- [1]Salciccioli KB; Zachariah JP Coarctation of the Aorta: Modern Paradigms Across the Lifespan. Hypertension, 2023.PMID 37476999
- [2]Kim YY; Andrade L; Cook SC Aortic Coarctation. Cardiol Clin, 2020.PMID 32622489
- [3]Hede SV; DeVore G; Satou G; et al Neonatal management of prenatally suspected coarctation of the aorta. Prenat Diagn, 2020.PMID 32277716
- [4]Bansal N; Balakrishnan PL; Aggarwal S Prostaglandin Infusion in Neonate With Severe Coarctation of the Aorta With Closed Ductus Arteriosus — A Case Report and Review of the Literature. World J Pediatr Congenit Heart Surg, 2020.PMID 31010402
- [5]LaPar DJ; Baird CW Surgical Considerations in Interrupted Aortic Arch. Semin Cardiothorac Vasc Anesth, 2018.PMID 29774793
- [6]Schreiber C; Mazzitelli D; Haehnel JC; et al The interrupted aortic arch: an overview after 20 years of surgical treatment. Eur J Cardiothorac Surg, 1997.PMID 9332928
- [7]Burbano-Vera N; Zaleski KL; Latham GJ; et al Perioperative and Anesthetic Considerations in Interrupted Aortic Arch. Semin Cardiothorac Vasc Anesth, 2018.PMID 29742969
- [8]Ron HA; Crowley TB; Liu Y; et al Improved Outcomes in Patients with 22q11.2 Deletion Syndrome and Diagnosis of Interrupted Aortic Arch Prior to Birth Hospital Discharge, a Retrospective Study. Genes (Basel), 2022.PMID 36672801
- [9]Eckhauser A; South ST; Meyers L; et al Turner Syndrome in Girls Presenting with Coarctation of the Aorta. J Pediatr, 2015.PMID 26323199
- [10]Wu Y; Jin X; Kuang H; et al Is balloon angioplasty superior to surgery in the treatment of paediatric native coarctation of the aorta: a systematic review and meta-analysis. Interact Cardiovasc Thorac Surg, 2019.PMID 30060099
- [11]Toro-Salazar OH; Steinberger J; Thomas W; et al Long-term follow-up of patients after coarctation of the aorta repair. Am J Cardiol, 2002.PMID 11867038
- [12]Sinning C; Zengin E; Kozlik-Feldmann R; et al Bicuspid aortic valve and aortic coarctation in congenital heart disease — important aspects for treatment with focus on aortic vasculopathy. Cardiovasc Diagn Ther, 2018.PMID 30740325