Paeds · cardiology
Congenital coronary anomalies and anomalous left coronary artery
Also known as ALCAPA · Bland-White-Garland syndrome · Anomalous left coronary artery from the pulmonary artery · Anomalous aortic origin of a coronary artery · AAOCA · Coronary artery fistula · Coronary artery anomaly
Fellowship guide to congenital coronary anomalies in children: the two-month-old with feeding-related pallor and irritability whose left coronary artery comes from the pulmonary artery (ALCAPA, Bland-White-Garland) and the outwardly well athlete whose coronary takes a malignant interarterial course (AAOCA) and collapses with exercise; the ECG anterolateral Q waves, echocardiographic definition of coronary origin and course, the ALCAPA steal, the surgical reimplantation that establishes dual coronary flow, risk stratification and exercise restriction for AAOCA, and the AATS expert consensus, ACC/AHA adult congenital guideline and athlete-screening positions.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the ten-week-old who screams and goes pale every time he feeds, then breathes fast and sweaty for an hour afterwards. His mother has been told it is colic, then reflux, then bronchiolitis. The electrocardiogram shows deep Q waves in the anterolateral leads that nobody expects in a baby, and the echocardiogram shows a big, floppy left ventricle that barely squeezes. The left coronary artery does not come from the aorta at all — it comes from the pulmonary artery. That is ALCAPA, and it is one of the few causes of infant heart failure that surgery can cure completely, which is why missing it is such a costly error. [1] [4]
Now picture the fifteen-year-old footballer who collapsed during a sprint and could not be resuscitated. He had been entirely well, and at autopsy his left coronary artery arose from the right sinus and ran a tight intramural course between the aorta and pulmonary artery. That is anomalous aortic origin of a coronary artery, and its first symptom is very often death. The thread connecting these two children is an outwardly normal heart whose coronary blood supply fails under a developmental or exertional load. [9] [13]
Congenital coronary anomalies are coronary arteries with an abnormal origin, course, number or termination present from birth. The great majority are incidental variants that never cause harm — a circumflex artery arising from the right sinus and passing behind the aorta, for example. The minority that matter do so because they reduce myocardial blood flow, either chronically, as in ALCAPA, or episodically with exertion, as in an interarterial AAOCA. The skill for a general paediatrician is to know the dangerous patterns and to prove the coronary origin and course with imaging whenever the heart is failing in infancy or symptoms occur with exercise. [2] [3]
Classification
The most useful way to classify congenital coronary anomalies is by what they do to myocardial blood flow, because that single axis drives urgency. A benign origin variant needs reassurance; a coronary that drains the left ventricle into the pulmonary artery needs an operating room. [1] [2]
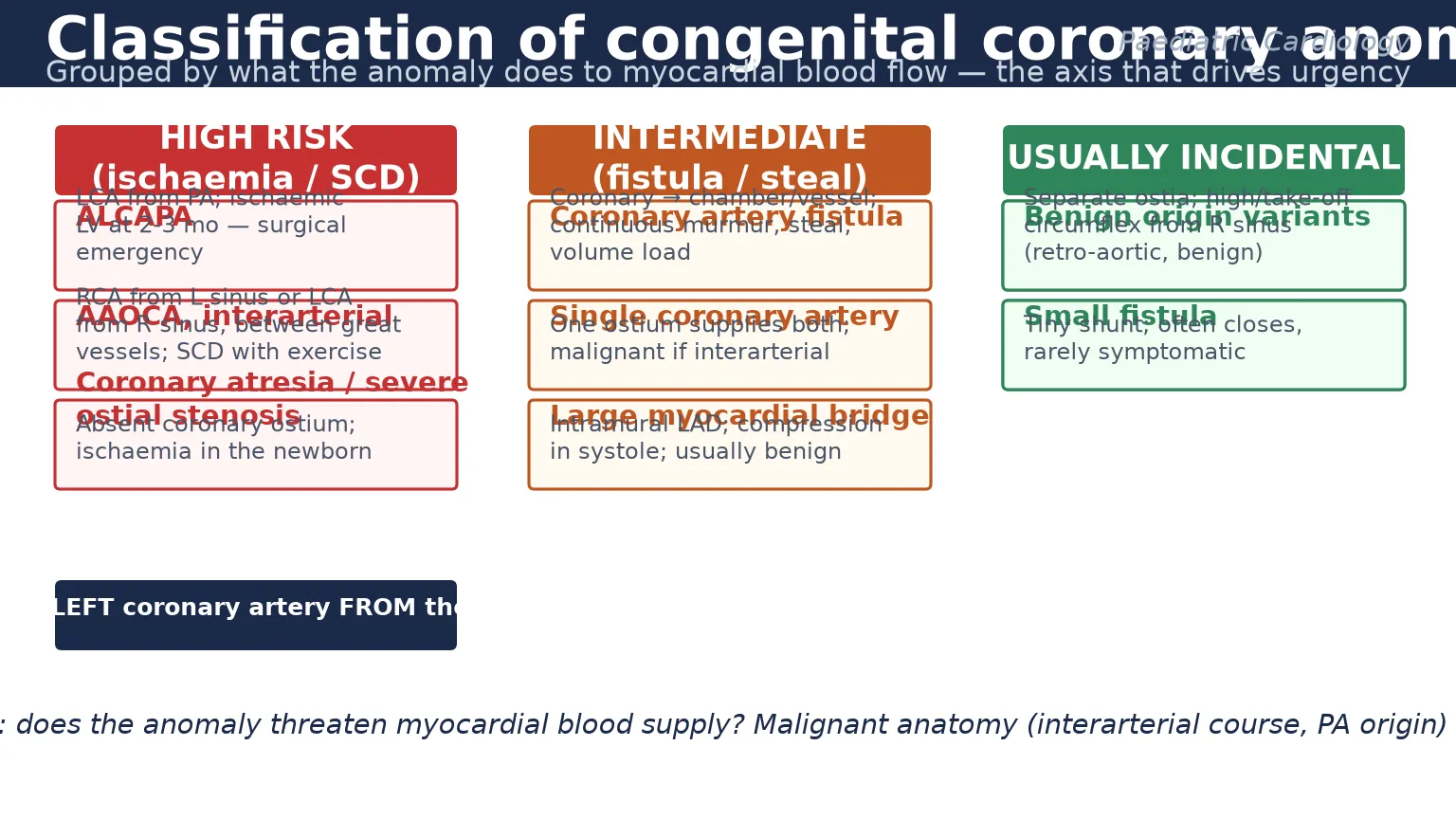
At the high-risk end sit three lesions. ALCAPA is the left coronary artery arising from the pulmonary artery and supplying the left ventricle with desaturated, low-pressure blood. Anomalous aortic origin with an interarterial course is a coronary from the opposite sinus that passes between the aorta and pulmonary artery, where the slit-like ostium and intramural segment pinch with exertion. Coronary atresia or severe ostial stenosis is an absent or critically narrowed coronary ostium that produces ischaemia in the newborn. Each of these can present as myocardial infarction, cardiogenic shock or sudden cardiac death. [9] [3]
The intermediate group includes coronary artery fistulae, where a coronary drains directly into a cardiac chamber or great vessel and produces a continuous murmur and a volume load; single coronary artery, where one ostium supplies the whole heart and is dangerous only if its branches run an interarterial course; and a large myocardial bridge, where a segment of coronary dives into the myocardium and is compressed in systole. These are watched, risk-stratified and treated only if they produce symptoms or a significant shunt. [1] [11]
ALCAPA
LCA from PA
- Infant 2–3 mo, feeding-related pallor
- Anterolateral Q waves on ECG
- Dilated poorly contracting LV
- Surgical emergency: dual coronary repair
AAOCA, interarterial
between great vessels
- Often asymptomatic until collapse
- Exertional chest pain/syncope
- LCA from R sinus = highest risk
- Restrict sport, image course, refer
Coronary fistula
coronary → chamber
- Continuous murmur in child
- Volume load, steal, heart failure
- Echo defines drainage site
- Close if large or symptomatic
Benign variant
incidental
- Separate ostia, high take-off
- Retro-aortic circumflex course
- No ischaemia or shunt
- Reassure, no restriction
The incidental group is the largest. Separate ostia for the left anterior descending and circumflex arteries, a high take-off above the sinus, and a circumflex artery from the right sinus passing behind the aorta are all normal-variant patterns with no haodynamic consequence. The trap for an examiner is to over-call these as dangerous when they are not, and the reverse trap is to miss a genuinely malignant interarterial course by assuming any odd-looking origin is benign. The decisive question is always whether the course passes between the great vessels. [2] [1]
Epidemiology & Risk Factors
Coronary anomalies are found in roughly one in three hundred people at coronary angiography or autopsy, and most are benign. ALCAPA is rare — about one in three hundred thousand live births, accounting for a quarter to a half of one per cent of all congenital heart disease — but it is over-represented in fellowship exams because it is a classic, curable, and frequently missed. Anomalous aortic origin is somewhat commoner, around one in one thousand to one in two thousand, and is a leading structural cause of sudden cardiac death in the young. [1] [9]
The risk profile is sharply different for the two dangerous lesions. ALCAPA is an equal-opportunity infant disease with a slight female predominance, and its risk is developmental — the symptoms appear as pulmonary vascular resistance falls over the first weeks of life. Anomalous aortic origin, by contrast, affects males more often than females, and its risk is exertional — the malignant anatomy only threatens myocardial flow under the cardiac output and aortic distension of competitive sport. [8] [13]
The numbers that anchor your viva
The strongest risk factors for a poor outcome are not inherited but situational. For ALCAPA, the risk is delay — the longer the myocardium is starved, the worse the ventricular function at repair and the lower the chance of full recovery. For anomalous aortic origin, the risk is sport — competitive athletics multiply the already-elevated risk of sudden death several-fold, which is the entire rationale for exercise restriction in those who are managed without surgery. [8] [10]
Pathophysiology
The central teaching in this topic is the ALCAPA steal, because it explains why an outwardly normal newborn becomes ischaemic at two to three months. In fetal life and the early neonatal period, the pulmonary vascular resistance is high and the pulmonary artery pressure is close to systemic. Blood therefore flows forward from the pulmonary artery into the misoriginated left coronary artery and perfuses the left ventricle, albeit with desaturated blood. The myocardium is protected. [1] [5]

Over the first weeks of life the pulmonary vascular resistance falls, as it should, and the pulmonary artery pressure drops well below systemic. Now the pressure gradient reverses. Blood flows from the left coronary artery, which is connected to the left ventricle, back into the low-pressure pulmonary artery rather than perfusing the myocardium. The left ventricle is left to be supplied only by collaterals from the right coronary artery, at low pressure and with desaturated blood. The myocardium infarcts, the ventricle dilates and fails, and the ischaemic papillary muscles produce mitral regurgitation. [1] [5]
The mechanism of ischaemia in anomalous aortic origin is different but the principle is the same — the supply fails under load. A coronary that arises from the opposite sinus and runs an interarterial course between the aorta and pulmonary artery often has a slit-like, tangential ostium and an intramural segment within the aortic wall. At rest the flow is adequate. Under the high cardiac output, tachycardia and aortic distension of competitive exercise, the intramural segment is compressed and the slit-like orifice deforms, so flow falls at the moment demand rises. The result is exertional ischaemia, malignant ventricular arrhythmia, and sudden cardiac death — frequently as the first and only symptom. [9] [8]
[8] [9]Clinical Presentation
ALCAPA announces itself in the second or third month of life with the anginal equivalents of infancy, and the skill is to recognise them as cardiac rather than gastrointestinal or respiratory. The infant becomes irritable, sweaty and pale during feeds, which are the effort-angina of the baby who has to suck. Parents describe crying that looks like pain, breathlessness, and poor feeding, followed by failure to thrive and then frank respiratory distress. The myocardial infarction is often silent in the history because infants cannot localise chest pain, so the presentation is of heart failure with a gallop, hepatomegaly and a mitral regurgitation murmur — or simply a shockingly unwell baby. [4] [5]
The electrocardiogram is the bedside clue that reorients the whole assessment. Infants with ALCAPA develop deep Q waves in the anterolateral leads — I, aVL and the left precordial leads — with ST-segment change and T-wave inversion, reflecting the anterolateral infarction. Q waves are not a normal infant finding in these leads, and their presence in a heart-failure baby should trigger an urgent search for the coronary origin. The chest radiograph shows cardiomegaly and pulmonary venous congestion, easily mistaken for pneumonia or bronchiolitis. [4]
Anomalous aortic origin is the opposite problem — it is usually silent until it is catastrophic. Most affected children are asymptomatic, and the anomaly is found incidentally on an echocardiogram done for another reason, or on screening, or at autopsy. When symptoms do occur they are exertional and should be treated as a red flag: chest pain, palpitation, dizziness or syncope during or immediately after exercise. Any of these in a young athlete demands that the coronary origin and course be proven before return to play. A continuous murmur, by contrast, points instead to a coronary artery fistula. [9] [13]
How the two dangerous anomalies present
Differential Diagnosis
For the infant with ALCAPA, the differential is the list of things that cause heart failure and a dilated left ventricle in early infancy, and the trap is to settle on one of them without checking the coronary origin. Dilated cardiomyopathy of metabolic, genetic or post-viral cause sits at the top, and the two are indistinguishable on symptoms alone — only the echocardiographic demonstration of the left coronary origin separates them. Myocarditis produces a similar big floppy ventricle with a viral prodrome, and septic shock with poor perfusion is the other acute mimic. Bronchiolitis, gastro-oesophageal reflux and infantile colic are the benign mimics that delay diagnosis, because the feeding-related pallor is misread as colic and the tachypnoea as lower-lobe pneumonia. [2] [5]
Dilated cardiomyopathy
the chief mimic
- Identical big floppy LV on echo
- Must show LCA comes from the aorta
- Metabolic, genetic or post-viral
- Medical management if coronaries normal
Myocarditis
viral prodrome
- Tachycardia, gallop, raised troponin
- Global dysfunction, normal coronaries
- Viral PCR positive
- Supportive, most recover
Bronchiolitis / colic
benign mimic
- Feeding pallor misread as colic
- Tachypnoea misread as pneumonia
- Cardiomegaly is the tell
- ECG Q waves are not explained
Sepsis
shock mimic
- Poor perfusion, hypotension
- Blood cultures, lactate
- Heart failure signs coexist
- Sepsis plus a big LV needs the coronary
For the young athlete, the differential of exertional syncope or sudden death is the cardiomyopathy and channelopathy family, and anomalous aortic origin sits squarely within it. Hypertrophic cardiomyopathy is the commonest structural cause of sudden death in the young athlete and produces a dynamic left ventricular outflow obstruction and an abnormal electrocardiogram. Arrhythmogenic right ventricular cardiomyopathy, long QT syndrome and catecholaminergic polymorphic ventricular tachycardia are the electrical causes, and aortic root disease such as Marfan syndrome with dissection completes the structural list. The unifying principle is that exertional syncope is never benign, and the work-up must include an electrocardiogram, echocardiogram with explicit coronary origin assessment, and exercise testing. [13] [9]
Clinical & Bedside Assessment
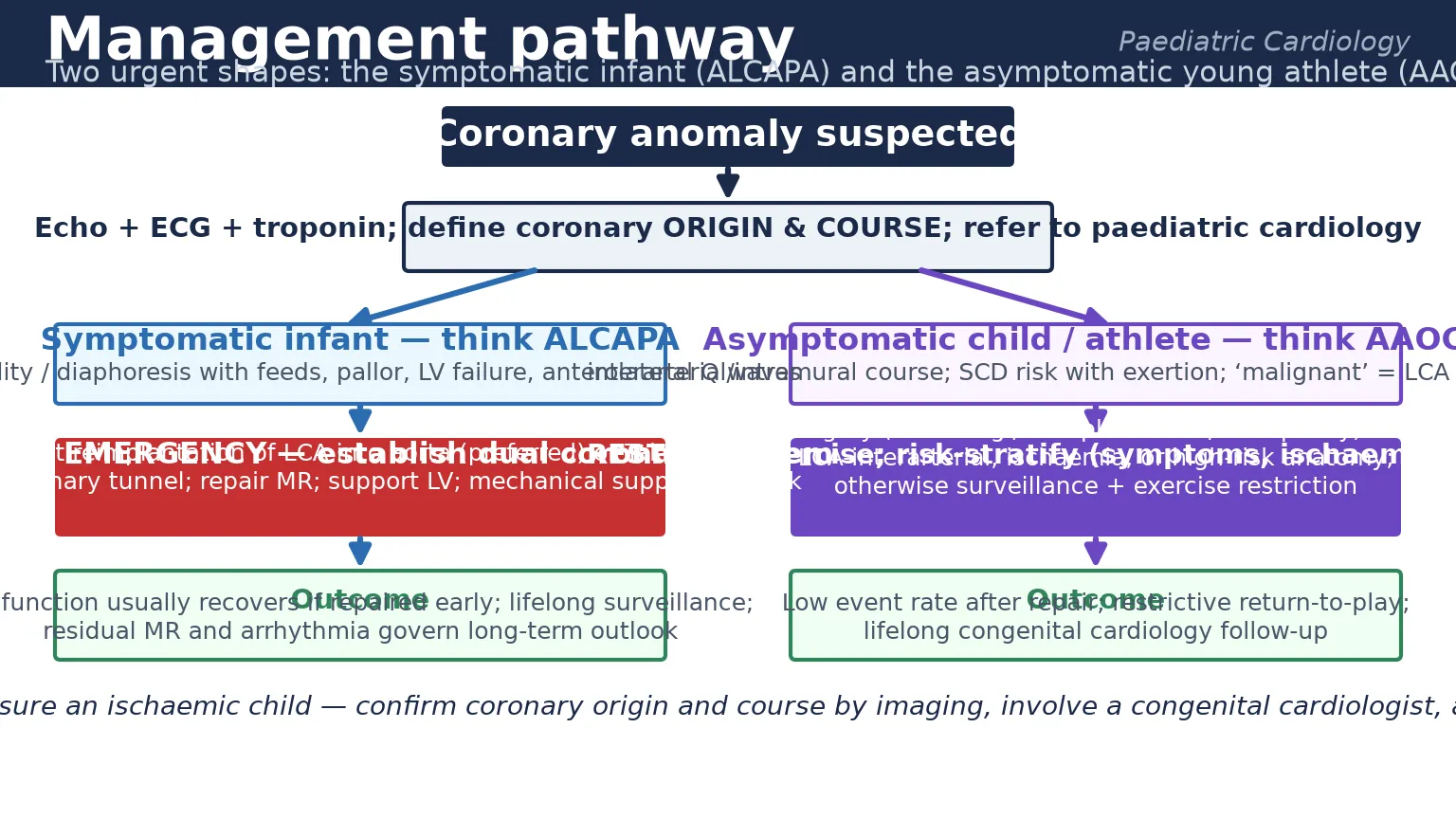
The bedside assessment begins with the recognition that an infant in heart failure or a young athlete with exertional symptoms carries a coronary anomaly until imaging proves otherwise. Take a focused history of the feeding pattern in the infant — when the pallor and sweating occur, whether they are effort-related, and whether there has been weight loss — and of the exertional symptoms in the athlete, including the precise trigger, the activity level, and any family history of sudden unexplained death. [1] [13]
[1] [9]Examine for the signs of heart failure in the infant — tachypnoea, retractions, a gallop rhythm, hepatomegaly and poor perfusion — and for a mitral regurgitation murmur at the apex, which reflects papillary-muscle ischaemia. A continuous murmur under the left sternal edge or at the lower sternal border should raise coronary artery fistula rather than a patent ductus in a child beyond the neonatal period. Weigh the infant, because the weight drives drug doses and the surgical plan. In the athlete, the resting examination is often normal, which is precisely why a normal examination must not reassure when the history carries exertional red flags. [11] [5]
Investigations
The electrocardiogram is the first and most accessible test, and in ALCAPA it carries a near-diagnostic pattern. Look for deep Q waves in leads I, aVL and the left precordial leads (V5 to V7), with ST-segment depression or elevation and T-wave inversion reflecting the anterolateral infarction, and left ventricular hypertrophy or strain from the dilated ventricle. A troponin is usually elevated in the symptomatic infant and in the athlete with an ischaemic event. The chest radiograph shows cardiomegaly and pulmonary oedema in ALCAPA, and is typically normal in anomalous aortic origin. [4]
Echocardiography is the cornerstone and the test that must not be left incomplete. The echocardiographer has two jobs: to show the left ventricular size and function, the mitral valve, and any thrombus, and — crucially — to demonstrate the origin and proximal course of both coronary arteries from the correct aortic sinus. In ALCAPA, colour Doppler shows retrograde flow in the left coronary artery draining into the pulmonary artery, a dilated right coronary artery from collateralisation, and a dilated poorly contracting left ventricle with mitral regurgitation. In anomalous aortic origin, the echocardiogram defines which sinus each coronary arises from and whether the course is interarterial, though the intramural segment may need cross-sectional imaging to characterise. [12] [5]
When echocardiography is equivocal, cross-sectional imaging defines the course. Computed tomography coronary angiography offers high spatial resolution and a fast acquisition that suits children, and it is the modality of choice for confirming an interarterial or intramural course. Cardiac magnetic resonance adds functional and scar information without radiation. Invasive coronary angiography remains the historical gold standard and is used when the non-invasive imaging is inconclusive or when intervention is planned. The goal of imaging is always the same — to prove where each coronary comes from and where it goes. [2] [3]
Management — Resuscitation
The infant with ALCAPA often presents in or near cardiogenic shock, and resuscitation is a bridge to definitive surgery rather than a substitute for it. Support the airway and breathing with oxygen and positive-pressure ventilation if the work of breathing is high, and treat the heart failure with diuretics such as furosemide and an afterload reducer such as an angiotensin-converting-enzyme inhibitor, recognising that the failing ventricle is dependent on afterload reduction to maintain forward flow. [5]
The genuinely unwell infant with low cardiac output needs intensive care. Inotropes such as milrinone and low-dose adrenaline support the contractility and the afterload, and mechanical circulatory support with extracorporeal membrane oxygenation is used for the infant who cannot be stabilised on inotropes, both as a bridge to surgery and as a bridge to recovery of the stunned ventricle after repair. Arrhythmia and acute mitral regurgitation may need separate management, and a metabolic acidosis signals how decompensated the circulation has become. The principle is to restore the circulation well enough to take the child safely to the operating room. [5]
The athlete with anomalous aortic origin and exertional ischaemia needs a different but equally prompt response. Restrict exercise immediately, admit for monitoring if there is ongoing ischaemia or arrhythmia, give antiplatelet and anti-anginal therapy as guided by the congenital cardiologist, and arrange urgent risk stratification with the definitive imaging and exercise testing that will determine whether surgery is required. The single most important resuscitation decision is to stop competitive sport, because exertion is the trigger for sudden death. [8] [10]
Management — Definitive & Stepwise
The definitive treatment of ALCAPA is surgical, and the goal is to re-establish a normal dual coronary system so the left ventricle is perfused from the aorta. The preferred operation is direct reimplantation, in which the left coronary artery is detached from the pulmonary artery with a button of arterial wall and re-implanted into the aorta, restoring antegrade systemic perfusion. Where the anatomy does not allow direct reimplantation — for example when the coronary is deeply embedded in the pulmonary artery wall — the Takeuchi operation creates an aortopulmonary window and tunnels a flap of pulmonary artery wall to redirect aortic blood into the left coronary ostium. [6] [7]
The surgical and post-operative arc of ALCAPA
Medical stabilisation: diuretics, afterload reduction, inotropes, mechanical support if shock
Confirm anatomy: echocardiography showing LCA from PA, retrograde flow, dilated LV and MR
Surgery: direct reimplantation of LCA into aorta, or Takeuchi intrapulmonary tunnel if anatomy demands
Address mitral regurgitation: repair at the index operation if severe, defer if moderate to allow recovery
Post-operative support: allow stunned LV to recover; mechanical support and inotropes wean over days
Lifelong surveillance: LV function, residual MR, arrhythmia; structured transition to adult congenital care
Mitral regurgitation is addressed at the same operation if it is severe, but moderate regurgitation is often left alone because the ischaemic papillary muscles recover once coronary flow is restored, and the regurgitation improves. The European Congenital Heart Surgeons Association database and long-term follow-up studies show that left ventricular function recovers in most children when repair is timely, that late mortality is low, and that residual mitral regurgitation and arrhythmia are the main determinants of long-term quality of life. Delay matters: the earlier the repair, the better the recovery, which is why this is a surgical emergency rather than an elective procedure. [6] [7]
The management of anomalous aortic origin is driven by anatomy and symptoms, and the AATS expert consensus provides the framework. An anomalous left coronary artery from the right sinus with an interarterial course carries the highest risk and is generally referred for surgery regardless of symptoms, because an ischaemic event in that territory is usually fatal. Surgical options include unroofing, which opens the intramural segment to create a normal-orientation ostium; reimplantation; and ostioplasty. An anomalous right coronary from the left sinus is risk-stratified: surgery is recommended for those with symptoms or documented ischaemia, while asymptomatic children without ischaemia may be managed with exercise restriction and surveillance. [8] [10]
Coronary artery fistulae are closed when they are large, symptomatic, or cause a significant shunt, and the choice is between transcatheter device or coil occlusion and surgical ligation, depending on the anatomy and the drainage site. Small, incidentally found fistulae are often monitored and may close spontaneously. A large myocardial bridge causing objective ischaemia is managed with beta-blockade and, rarely, surgical unroofing; most bridges are incidental and need no treatment. [11] [1]
Specific Subtypes & Scenarios
ALCAPA is the subtype that dominates the infant presentation, and its two clinical shapes are worth holding separately. The classic infantile presentation is the two- to three-month-old with feeding-related angina and a dilated left ventricle, as described above. The rarer adult presentation occurs when extensive collaterals from the right coronary artery keep the left ventricle perfused into adulthood, and the patient presents with mitral regurgitation, ischaemia, or sudden death decades later. The difference is the collateral circulation, which is the single variable that decides whether ALCAPA declares itself in infancy or hides until adulthood. [6] [5]
Anomalous aortic origin splits along the artery and the course. An anomalous left coronary artery from the right sinus with an interarterial, intramural course is the malignant lesion — high sudden-death risk, low threshold for surgery. An anomalous right coronary artery from the left sinus carries a lower risk, and management is individualised to symptoms and ischaemia. The terms interarterial — passing between the aorta and pulmonary artery — and intramural — running within the aortic wall — describe the anatomy that matters, because both contribute to the dynamic obstruction that produces exertional ischaemia. [8] [10]
CORONARY
Coronary artery fistula is the continuous-murmur subtype. The fistula connects a coronary artery to a cardiac chamber — most often the right ventricle, right atrium or pulmonary artery — or to the venous system. A small fistula is asymptomatic; a large one produces a continuous murmur, a volume load on the receiving chamber, biventricular enlargement, and a steal phenomenon that can cause ischaemia in the parent coronary. Endocarditis and thrombosis within the fistula are recognised complications. Closure is by transcatheter device or surgery when the shunt is significant or the patient is symptomatic. [11]
The remaining subtypes are less common but examinable. Single coronary artery is dangerous only when one of its branches runs an interarterial course, when it behaves like anomalous aortic origin. Coronary atresia presents with ischaemia in the newborn and may be duct-dependent, requiring prostaglandin until surgical repair. A myocardial bridge is usually an incidental intramyocardial segment of the left anterior descending artery that is compressed in systole; it is benign unless it produces objective ischaemia, when beta-blockade or, rarely, surgical unroofing is considered. [1] [2]
Complications & Pitfalls
The defining complication of an unrecognised coronary anomaly is irreversible. In ALCAPA it is the anterolateral infarction that scars the left ventricle, leaves the child with chronic heart failure, and converts a curable lesion into a chronic cardiomyopathy or a transplant referral. In anomalous aortic origin it is sudden cardiac death, frequently without any prodrome, which is why the lesion is so often first diagnosed at autopsy. Mitral regurgitation, ventricular arrhythmia and progressive left ventricular dysfunction are the complications that dominate long-term follow-up after repair. [6] [9]
The avoidable pitfalls are several, and each is a classic examination trap. The first is treating ALCAPA as idiopathic dilated cardiomyopathy without ever showing that the left coronary artery comes from the aorta — the echocardiographer must be asked the direct question, and the coronaries must be seen. The second is dismissing an infant's feeding-related pallor as colic and the tachypnoea as bronchiolitis, when the cardiomegaly and anterolateral Q waves are staring from the investigations. The third is reassuring a young athlete with exertional syncope, because a normal resting electrocardiogram and a normal examination do not exclude an anomalous coronary. The fourth is over-calling a benign retro-aortic circumflex variant as dangerous, or missing a malignant interarterial course by assuming that any unusual origin is benign. [3] [8]
[13]Prognosis & Disposition
The prognosis of ALCAPA after timely surgical repair is excellent and is the single most encouraging fact in the topic. Left ventricular function recovers in most children when the repair is done before irreversible scarring, the mitral regurgitation often regresses once coronary flow is restored, and late mortality is low. The determinants of a poorer outcome are delayed repair with established infarction, severe residual mitral regurgitation, and ventricular arrhythmia from scar. These children enter lifelong surveillance with serial echocardiography and a structured transition to adult congenital cardiac care in adolescence. [6] [7]
The prognosis of anomalous aortic origin depends on the artery, the course and the management. After successful unroofing or reimplantation, the event rate is low and many athletes return to competitive sport under a structured, supervised protocol. For those managed without surgery, exercise restriction and surveillance carry a small but real residual risk, which is the trade-off that families and clinicians must weigh. The disposition in every case is into a congenital cardiology service with expertise in coronary anomalies, because the long-term decisions about sport, imaging and reoperation are subspecialty decisions. [10] [8]
Coronary fistulae that are closed have a good prognosis, with a small risk of recurrence or residual flow. Single coronary artery and coronary atresia have outcomes driven by the associated anatomy and the success of surgical repair, and a myocardial bridge that is truly incidental carries no excess risk. Across all subtypes, the disposition principle is the same: a coronary anomaly that threatens blood flow needs a congenital cardiologist for life, and one that does not needs only clear reassurance. [2] [11]
Special Populations
Infants dominate the ALCAPA population, and the special consideration in this group is the subtlety of the symptoms. A baby cannot describe angina, so the clinician must read the feeding-related pallor, diaphoresis and irritability as the anginal equivalent, and must treat the dilated left ventricle as a coronary anomaly until imaging proves otherwise. The threshold to image the coronary origin in any infant with unexplained heart failure is therefore very low. [5] [4]
Adolescents and young athletes dominate the anomalous aortic origin population, and the special consideration is the interaction with competitive sport. The young athlete values participation highly, and exercise restriction carries a real psychological and social cost, so the decision to operate, restrict or clear must be shared, evidence-based, and made with a congenital cardiologist. Structured return-to-play protocols after surgery allow many athletes to compete again under surveillance. The transition to adult congenital care is a second special-population task, because these patients need lifelong follow-up that the general adult system is not equipped to provide. [10]
Families matter in two further ways. A young sudden cardiac death prompts evaluation of first-degree relatives for inheritable cardiomyopathy and channelopathy, and although most coronary anomalies are sporadic rather than inherited, the family screening that follows a death is an opportunity to detect other at-risk young people. Families also carry the psychological burden of an exercise restriction or a major cardiac surgery in a child, and they need honest communication, a clear surveillance plan, and a named point of contact in the congenital service. [13]
Evidence, Guidelines & Regional Differences
The evidence base for congenital coronary anomalies combines expert consensus, large surgical registries and observational data, because randomised trials are impractical in rare lesions. The AATS expert consensus guidelines on anomalous aortic origin of a coronary artery, published in the Journal of Thoracic and Cardiovascular Surgery, set the framework for risk stratification and the surgical threshold, distinguishing the high-risk left-sided interarterial anomaly from the individualised right-sided lesion. The ACC/AHA guideline for the management of adults with congenital heart disease extends the surveillance and transition principles into adult life. [8] [3]
The European Congenital Heart Surgeons Association database has reported surgical outcomes for ALCAPA repair across many centres, confirming that direct reimplantation is the preferred technique and that early repair yields good long-term left ventricular recovery. Long-term single-centre follow-up studies have refined the understanding of residual mitral regurgitation and arrhythmia, and have shifted practice toward earlier repair and selective mitral intervention. Imaging reviews have established computed tomography and echocardiography as the complementary modalities that define the origin and course without routine invasive angiography. [6] [7]
How the guidelines and consensus documents fit together
The main regional differences concern screening and access. The Italian electrocardiographic pre-participation screening programme, endorsed by the European Society of Cardiology, has demonstrated a fall in sudden cardiac death in young athletes, and the screening debate turns on the trade-off between lives saved and the false-positive rate and cost. In Australia and New Zealand, the retrieval and telehealth network supports rural and remote clinicians in stabilising and transferring the infant with ALCAPA, and practice aligns with the AATS consensus and the ACC/AHA guideline. In lower-resource settings, late presentation with established ventricular damage and limited access to infant cardiac surgery remain the dominant realities. [13] [9]
Exam Pearls
[1] [8] [6]References
- [1]Gentile F, Castiglione V, De Caterina R Coronary Artery Anomalies. Circulation, 2021.PMID 34543069
- [2]Evangelista M, Ferrero P, D'Aiello AF, Negura D Coronary artery anomalies: what are they? when to suspect? how to treat?-a narrative review. Transl Pediatr, 2024.PMID 39144437
- [3]Chandrasekhar S, Woods E, Bennett J, Newman N Coronary Artery Anomalies: Diagnosis & Management. Cardiol Rev, 2024.PMID 39315746
- [4]Hoffman JI Electrocardiogram of anomalous left coronary artery from the pulmonary artery in infants. Pediatr Cardiol, 2013.PMID 23242106
- [5]Cashen K, Kwiatkowski DM, Riley CM, Buckley J Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery: A Retrospective Multicenter Study. Pediatr Crit Care Med, 2021.PMID 34432672
- [6]Thomas AS, Chan A, Alsoufi B, Vinocur JM Long-term Outcomes of Children Operated on for Anomalous Left Coronary Artery From the Pulmonary Artery. Ann Thorac Surg, 2022.PMID 34419434
- [7]Triglia LT, Guariento A, Zanotto L, Zanotto L Anomalous left coronary artery from pulmonary artery repair: Outcomes from the European Congenital Heart Surgeons Association Database. J Card Surg, 2021.PMID 33651393
- [8]Brothers JA, Frommelt MA, Jaquiss RDB, Myerburg RJ Expert consensus guidelines: Anomalous aortic origin of a coronary artery. J Thorac Cardiovasc Surg, 2017.PMID 28274557
- [9]Schiavone M, Gobbi C, Gasperetti A, Zuffi A Congenital Coronary Artery Anomalies and Sudden Cardiac Death. Pediatr Cardiol, 2021.PMID 34459947
- [10]Molossi S, Sachdeva S Advice to Young Athletes With Anomalous Aortic Origin of a Coronary Artery With and Without Surgery. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu, 2025.PMID 40382130
- [11]Goo HW Imaging Findings of Coronary Artery Fistula in Children: A Pictorial Review. Korean J Radiol, 2021.PMID 34564965
- [12]Brown LM, Duffy CE, Mitchell C, Young L A practical guide to pediatric coronary artery imaging with echocardiography. J Am Soc Echocardiogr, 2015.PMID 25691000
- [13]Corrado D, Zorzi A Sudden death in athletes. Int J Cardiol, 2017.PMID 28318658