Paeds · cardiology
Duct-dependent congenital heart disease: recognition and stabilisation
Also known as Duct-dependent congenital heart disease · Critical congenital heart disease · Duct-dependent circulation · Neonatal cardiac collapse · Duct-dependent pulmonary circulation · Duct-dependent systemic circulation · Prostaglandin E1 therapy · Critical CHD
Fellowship guide to duct-dependent congenital heart disease in neonates and infants: the two-pathway split (duct-dependent pulmonary versus systemic circulation), the physiology of why a baby who was well on day one collapses on day three, the hyperoxia test and pulse oximetry screening, emergency prostaglandin E1 (alprostadil) therapy with its apnoea and hypotension pitfalls, and the transfer pathway to a cardiac surgical centre.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The organising principle is simple: the ductus arteriosus is a bridge, and duct-dependent lesions are the ones where that bridge is the only road. In fetal life the placenta oxygenates the blood and the ductus arteriosus shunts most of the right ventricular output away from the lungs. After birth the lungs take over, oxygen tension rises, and the duct closes functionally within hours and anatomically over days. If the lesion blocks the pulmonary outflow, closing the duct cuts the only route blood had to reach the lungs, and the baby turns blue. If the lesion blocks the systemic outflow, closing the duct cuts the only route to the body, and the baby goes into shock. [3]
This page covers the recognition and emergency stabilisation of duct-dependent congenital heart disease in neonates and infants. It works through the two-pathway split — duct-dependent pulmonary versus duct-dependent systemic circulation — the physiology of why a well baby collapses at day three, the pulse oximetry screening programme and the hyperoxia test, the emergency prostaglandin E1 protocol with its apnoea and hypotension traps, the parallel sepsis workup, and the retrieval pathway to a cardiac surgical centre. It links to the murmur assessment, shock, and neonatal sepsis topics rather than repeating their full detail. [4]
Overview & Definition
A baby born with a severe obstruction on one side of the heart can survive in fetal life because the ductus arteriosus lets blood detour around the block. The condition earns its name — duct-dependent — because that detour is not a luxury but a lifeline. The moment the duct closes, the child loses the only route for either pulmonary or systemic blood flow, and the clinical picture is either deep cyanosis or cardiovascular collapse. [3]
Duct-dependent pulmonary circulation covers the right-sided obstructions where blood cannot reach the lungs once the duct closes: pulmonary atresia, critical pulmonary stenosis, severe tetralogy of Fallot, tricuspid atresia, and severe Ebstein anomaly. Before the duct closes, blood flows from the aorta through the ductus into the pulmonary artery and reaches the lungs in reverse. When the duct shuts, pulmonary blood flow plummets and the baby becomes deeply cyanotic. [3]
Duct-dependent systemic circulation covers the left-sided obstructions where blood cannot reach the body once the duct closes: hypoplastic left heart syndrome, critical coarctation of the aorta, interrupted aortic arch, critical aortic stenosis, and mitral atresia. Before the duct closes, the right ventricle pumps blood through the ductus into the aorta and perfuses the body. When the duct shuts, systemic output collapses and the baby presents in shock with weak pulses and metabolic acidosis. [3]
Classification
The fastest way to classify these lesions is by which circulation fails when the duct closes, because that decides the presentation, the physical signs, and the initial resuscitation priority. The figure below splits the two pathways and lists the lesions that sit on each. [3]

Duct-dependent pulmonary
- Right-sided obstruction: blood cannot reach the lungs
- Lesions: pulmonary atresia, critical pulmonary stenosis, severe TOF, tricuspid atresia, Ebstein
- Presents with CYANOSIS unresponsive to oxygen
- Typically deep desaturation from birth or worsening over days
Duct-dependent systemic
- Left-sided obstruction: blood cannot reach the body
- Lesions: HLHS, critical coarctation, interrupted aortic arch, critical aortic stenosis
- Presents with SHOCK, acidosis, weak or absent femoral pulses
- May be pink initially, then collapses on day 2 to 5
Congenital heart disease is the commonest birth defect, occurring in about nine per thousand live births, and the worldwide meta-analysis by van der Linde and colleagues confirmed that this rate holds across populations. About a quarter of these lesions are critical, meaning they need surgery or catheter intervention in the first year, and a substantial fraction of those are duct-dependent. Hoffman and Kaplan established the modern incidence benchmark, and their data underpin every screening and service-planning decision that follows. [1] [2]
Epidemiology & Risk Factors
The epidemiology of duct-dependent lesions is the epidemiology of congenital heart disease filtered for severity. Roughly one in four children born with structural heart disease has a lesion that is critical, and a meaningful proportion of those — including all the hypoplastic left heart, pulmonary atresia, and critical coarctation cases — depend on a patent ductus to survive the neonatal period. [1] [2]
The dominant risk factor for collapse at home is missed diagnosis. Wren and colleagues showed in their twenty-year UK study that despite advances in antenatal ultrasound and postnatal examination, a significant proportion of life-threatening cardiovascular malformations were still not diagnosed before discharge from hospital, and the duct-dependent left-sided obstructions were the most frequently missed because the baby looks pink and well on the first day. The risk of a missed duct-dependent lesion falls sharply when pulse oximetry screening is added to routine newborn examination. [9]
DUCTS
A well baby collapsing at day 2 to 5 is the classic duct-dependent presentation
Cyanosis not correcting with 100% oxygen suggests a cardiac cause
Absent femoral pulses point to coarctation or interrupted aortic arch
Prostaglandin E1 at 0.01 to 0.05 micrograms per kilogram per minute reopens the duct
Sepsis mimics and coexists with duct-dependent CHD — treat both in parallel
Risk factors for having a critical congenital heart lesion include a family history of CHD, maternal diabetes, certain teratogenic exposures, and chromosomal syndromes such as Turner, Noonan, and 22q11 deletion. But most duct-dependent lesions occur in babies with no identifiable risk factor, which is why universal screening rather than targeted screening is the standard. [3]
Pathophysiology
In fetal life the placenta provides oxygen and the lungs are bypassed. The ductus arteriosus carries most of the right ventricular output from the pulmonary artery directly into the aorta, so the high-resistance lungs receive only a small fraction of combined cardiac output. Blood then returns to the placenta via the umbilical arteries, and oxygenation happens outside the body. [3]
At birth three things happen in rapid succession. The cord is clamped and the low-resistance placental circuit is removed, raising systemic vascular resistance. The lungs expand, dropping pulmonary vascular resistance. And oxygen tension in the blood rises sharply. These changes reverse the pressure gradient across the ductus arteriosus, so flow through the duct changes from right-to-left (pulmonary to systemic) to left-to-right, and then stops. Rising oxygen tension and falling prostaglandin levels cause the duct to close functionally within hours and anatomically over the following days. [3]
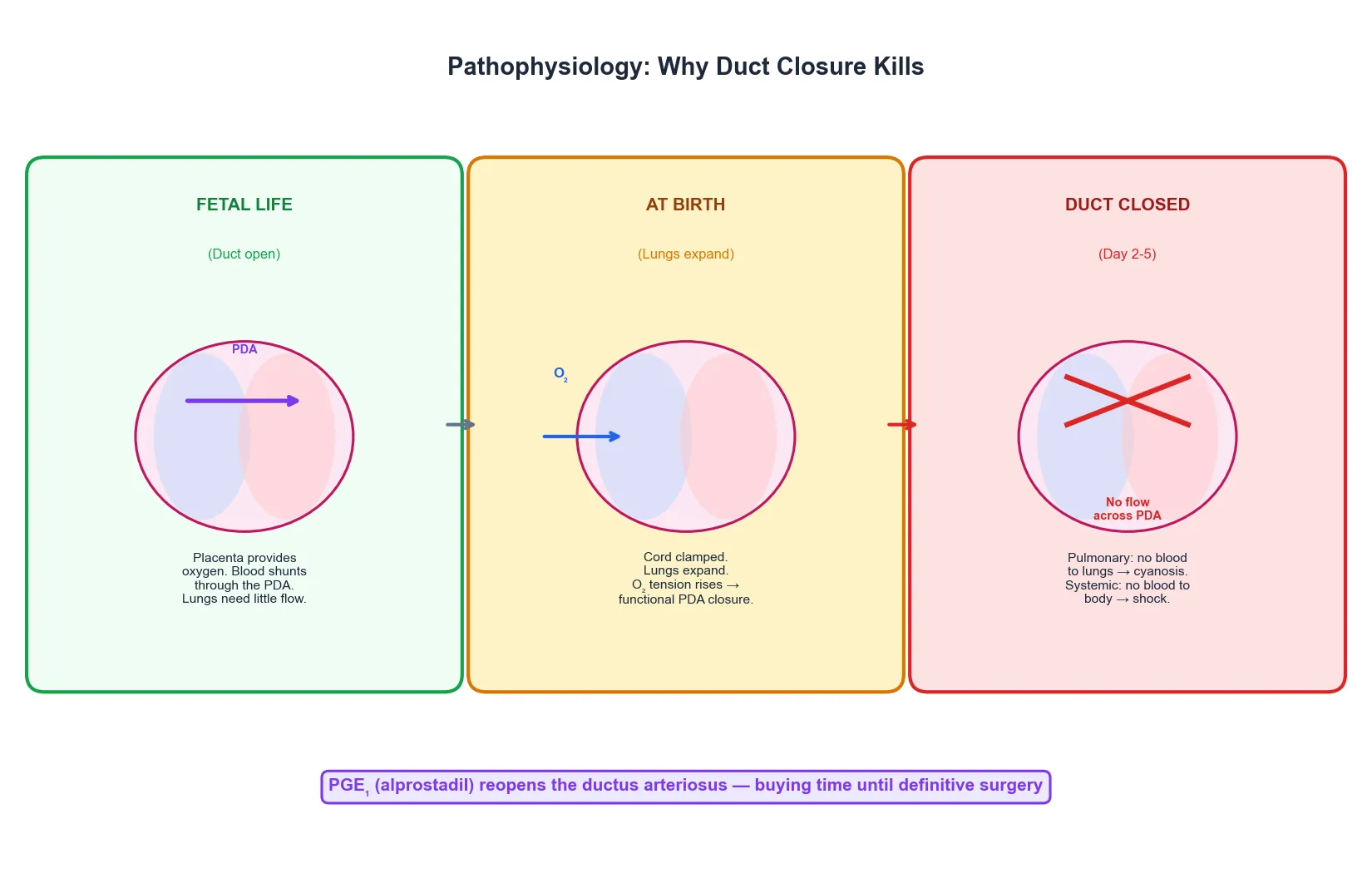
In a baby with a normal heart this is uneventful because the lungs and the systemic circulation can each handle their full share of cardiac output through their own pathways. In a baby with a duct-dependent lesion, the ductus arteriosus was the only detour around the obstruction, and closing it removes the only route. In duct-dependent pulmonary circulation, blood can no longer reach the lungs and the baby becomes cyanotic because the pulmonary blood flow that was coming retrograde through the duct has stopped. In duct-dependent systemic circulation, blood can no longer reach the body and the baby collapses in shock because the systemic output that was coming antegrade through the duct has stopped. [3] [11]
This is why the timing of collapse is so predictable. The duct closes functionally within the first day but can maintain a trickle of flow until it closes anatomically at day two to five. The baby who was discharged home looking well returns to the emergency department on day three or four in extremis, and the history of a preceding normal period is the clue that distinguishes this from early-onset sepsis or a metabolic crisis. [9]
Clinical Presentation
Duct-dependent lesions present in two patterns that mirror the two pathways. The cyanotic pattern is a baby who is blue from birth or who becomes progressively more cyanotic over the first days, with a saturation that does not improve when oxygen is given. The shock pattern is a baby who looks pink and well initially, then collapses on day two to five with poor perfusion, tachypnoea, grunting, poor feeding, lethargy, and weak or absent pulses. Both can present with apnoea, respiratory distress, or cardiogenic shock as the duct closes further. [3] [11]
The cyanotic neonate with a duct-dependent pulmonary lesion may have an audible murmur — a harsh systolic murmur of right ventricular outflow obstruction, or a single second heart sound — but many critical lesions have no murmur because there is no significant turbulent flow across the obstruction. The absence of a murmur never excludes critical CHD. Look for differential cyanosis, where the hands are pink but the feet are blue, which suggests a right-to-left ductal shunt feeding the lower body with desaturated blood while the upper body receives well-oxygenated blood. [3]
The shocked neonate with a duct-dependent systemic lesion classically has weak or absent femoral pulses, a finding that is easy to miss if the pulses are not deliberately checked in all four limbs. Blood pressure in the arms may be normal or high while the legs are hypotensive, and a differential of more than 20 millimetres of mercury between upper and lower limb systolic pressures is highly suggestive of coarctation. The baby may have hepatomegaly from venous congestion, a gallop rhythm, and signs of poor perfusion such as mottled skin, prolonged capillary refill, and cool extremities. [3]
Differential Diagnosis
The differential of a neonate presenting with cyanosis or shock on day two to five is broad, and the three conditions that most closely mimic duct-dependent CHD are sepsis, persistent pulmonary hypertension of the newborn, and congenital adrenal hyperplasia. All four can present with respiratory distress, shock, and acidaemia, and they can coexist, which is why the initial workup casts a wide net and treatment runs in parallel rather than waiting for a single diagnosis. [3]
Sepsis is the default differential and must always be covered. A neonate with duct-dependent CHD can develop secondary sepsis from poor perfusion and gut ischaemia, and a neonate with sepsis can have a saturation that looks like a cardiac lesion. The distinguishing features are the saturation response to oxygen and the timing. In sepsis the saturation usually improves with oxygen; in duct-dependent pulmonary disease it does not. And sepsis can present at any time, while the classic duct-dependent collapse clusters at day two to five. Cover with cultures and empiric antibiotics in every case. [11]
Persistent pulmonary hypertension of the newborn presents with cyanosis that is often differential, and like duct-dependent CHD the saturation may not improve with oxygen. The distinguishing test is the hyperoxia test combined with pre- and post-ductal saturation comparison, and the definitive separator is echocardiography. Congenital adrenal hyperplasia presents with vomiting, shock, and hyponatraemia with hyperkalaemia in a phenotypically female baby, and the electrolyte pattern and the 17-hydroxyprogesterone distinguish it. Metabolic disease presenting with collapse, respiratory alkalosis, or metabolic acidosis is another parallel diagnosis to consider, especially if there is hypoglycaemia or hyperammonaemia. [3]
Clinical & Bedside Assessment
The bedside assessment of a suspected duct-dependent neonate runs on two tracks at once. The first is the immediate ABCDE assessment to judge how sick the child is and whether you need to resuscitate before you investigate. The second is the focused cardiac examination that gathers the clues to whether the problem is pulmonary or systemic, and whether it is cardiac at all. Check the airway, assess the work of breathing and the oxygen saturation, feel the pulses in all four limbs, measure the blood pressure, and assess perfusion with capillary refill and skin temperature. [3]
The bedside glucose is essential because hypoglycaemia is common in the shocked neonate and is both a consequence and a contributor to the collapse. A venous or capillary blood gas gives the pH, the carbon dioxide, the base deficit, and the lactate, and the degree of metabolic acidosis is a direct measure of how badly the tissues are being underperfused. In the shocked duct-dependent baby the acidosis is often severe, and correcting it with fluids alone is impossible because the underlying problem is obstructed blood flow, not volume depletion. [11]
The focused cardiac examination looks for the murmur, the heart sounds, and the signs of heart failure. Listen for a systolic murmur of outflow tract obstruction, a single or narrowly split second heart sound suggesting pulmonary hypertension or a great-vessel anomaly, and a gallop rhythm suggesting ventricular dysfunction. Examine the abdomen for hepatomegaly, which is the sign of right heart failure or venous congestion in the infant. Look at the colour pattern: is the cyanosis central, differential, or acrocyanotic? Assess the precordium for a thrill or a hyperdynamic impulse. [3]
Investigations
The investigation plan answers three questions: is this cardiac, which side is blocked, and what else might be going on. The first-line tests are done in parallel with resuscitation and do not delay the start of prostaglandin E1. The echocardiogram is the definitive test but it confirms rather than initiates treatment. [4]
The hyperoxia test is the classical bedside discriminator between cardiac and pulmonary cyanosis. Place the baby in 100 percent oxygen for ten minutes and measure an arterial partial pressure of oxygen. A PaO2 above 100 millimetres of mercury effectively excludes a right-to-left shunt and points to a pulmonary cause, while a PaO2 below 100 suggests a fixed right-to-left cardiac shunt that cannot be overcome by oxygen. The test has limitations: it is unreliable in pulmonary hypertension, it can be dangerous if the baby is already unstable, and the saturation response to oxygen is often enough to make the clinical decision without a formal PaO2. [3]
Pulse oximetry screening is the public health answer to the missed duct-dependent lesion. The 2012 Lancet meta-analysis by Thangaratinam and colleagues pooled data from nearly a quarter of a million babies and showed that pulse oximetry alone detected about three-quarters of critical congenital heart defects, with a false positive rate under one percent, and that adding it to routine newborn examination increased overall detection to over ninety percent. The American Heart Association and American Academy of Pediatrics joint statement, led by Mahle, codified the screening protocol: measure in the right hand (pre-ductal) and either foot (post-ductal) after 24 hours of age, and fail the screen if either is below 90 percent, if either is below 95 percent on three repeated measures, or if there is a differential of more than 3 percent. [4] [7]
The echocardiogram is the definitive investigation and should be performed as soon as the baby is stabilised on prostaglandin E1. It defines the anatomy, the direction and size of the ductal shunt, the ventricular function, and any associated lesions. In centres without on-site paediatric echocardiography, a telemedicine link to a cardiac centre can guide the prostaglandin dose and the timing of transfer. The echo confirms but does not initiate treatment — if the child is sick and the suspicion is high, start the prostaglandin first. [8]
Management — Resuscitation
The resuscitation of duct-dependent CHD is built around one drug — prostaglandin E1 (alprostadil) — given as early as possible to reopen or maintain the ductus arteriosus. Everything else supports that single intervention: secure the airway, gain vascular access, correct the glucose and acid-base disturbance, cover sepsis, and arrange transfer. The figure below shows the full algorithm. [10]
Start prostaglandin E1 (alprostadil) intravenously at 0.01 micrograms per kilogram per minute and titrate up to 0.05 micrograms per kilogram per minute until the saturation improves or the perfusion corrects. The infusion works within minutes to hours by relaxing the smooth muscle of the ductus arteriosus and restoring the shunt. In a cyanotic baby you will see the saturation rise; in a shocked baby you will see the perfusion, the acidosis, and the pulses improve. Do not exceed the minimum effective dose, because the side effects — apnoea, hypotension, fever, and diarrhoea — are dose-dependent. [10]
Across the American Heart Association, the European Society of Cardiology, and the Royal Australasian College of Physicians guidelines, the acute management of suspected duct-dependent CHD is identical: start prostaglandin E1 on clinical suspicion, intubate if the baby is apnoeic or needs transport, cover sepsis in parallel, and transfer to a cardiac surgical centre for definitive repair or palliation. Local protocols differ on the starting dose and on whether to electively intubate for transport, but the principle — reopen the duct first, confirm the diagnosis second — is universal. [4] [8]
Anticipate the side effects of prostaglandin E1, because they are common and can be dangerous. Apnoea occurs in roughly one in ten babies on the infusion and is the reason many units electively intubate before transfer. Hypotension, fever, and flushing are vasodilatory effects that usually respond to dose reduction or fluid. Pyrexia can mask a fever from sepsis, so do not attribute fever to the prostaglandin without sending cultures. Lewis and colleagues established the side-effect profile in their landmark Circulation paper, and the message has not changed: the drug is life-saving but it requires airway and blood-pressure vigilance. [10]
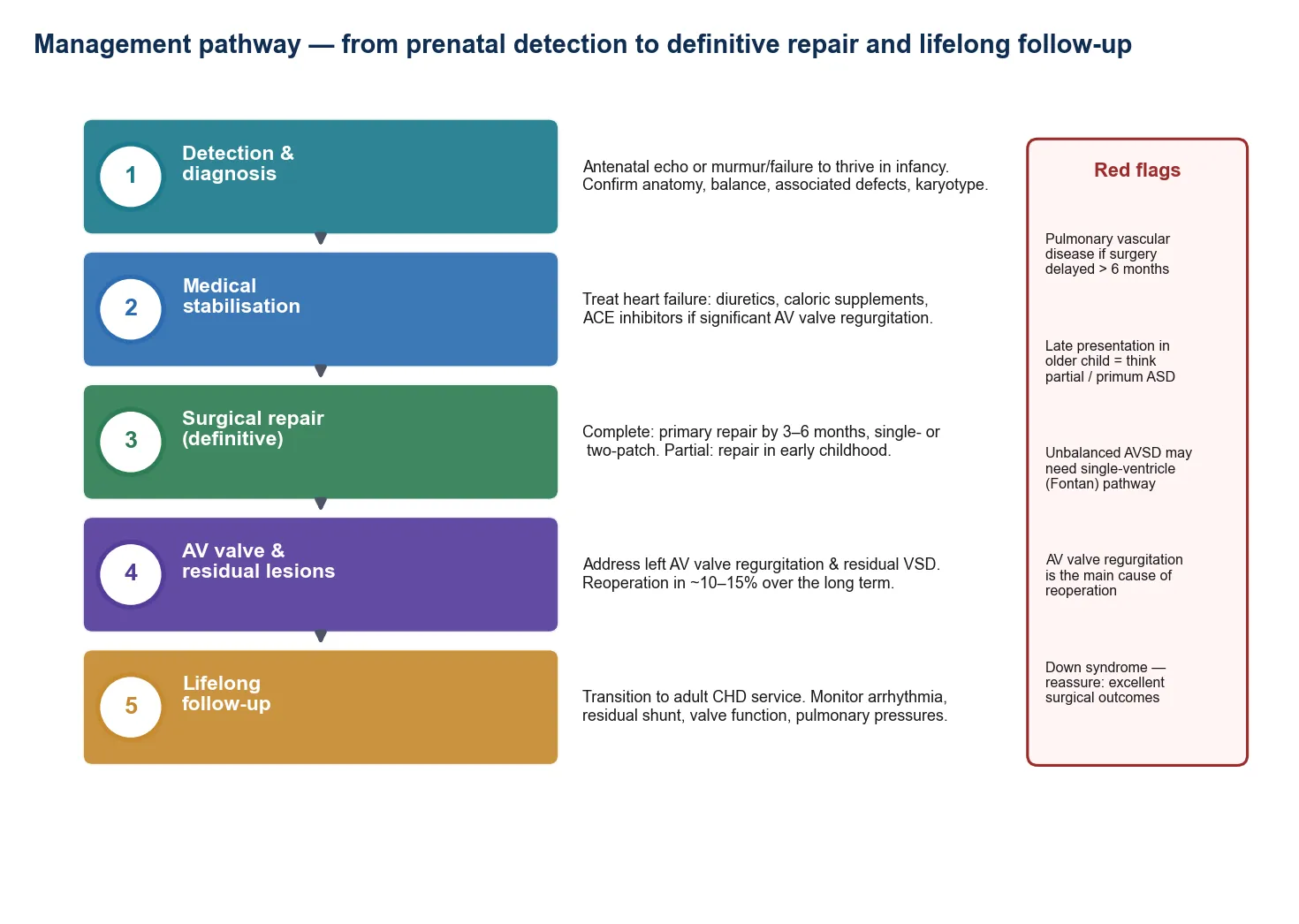
Management — Definitive & Stepwise
Once the duct is open and the baby is stable, definitive management is surgical and happens at a cardiac centre. The general paediatrician or neonatologist's job is to maintain the prostaglandin infusion, keep the baby physiologically stable, and effect a safe transfer. The definitive surgery depends on the lesion, and ranges from a single-stage repair — such as an arterial switch for transposition — to a staged palliation over the first years of life for single-ventricle lesions such as hypoplastic left heart syndrome. [3]
The definitive pathway, from collapse to surgery
Recognise the collapse and start prostaglandin E1 on clinical suspicion
Stabilise: airway, ventilation if apnoeic, correct glucose and acidosis, cover sepsis
Confirm anatomy with echocardiography (paediatric cardiology, on-site or telemedicine)
Maintain prostaglandin infusion at the minimum effective dose; monitor for apnoea and hypotension
Plan retrieval: dedicated neonatal transport team, intubate if transferring on prostaglandin
Transfer to a cardiac surgical centre for definitive repair or staged palliation
Long-term follow-up: cardiac surgery, neurodevelopmental surveillance, and structured transition
The prostaglandin infusion continues from the moment it is started until the definitive surgery is undertaken, which may be hours to days depending on the lesion, the centre, and the transfer logistics. During this period the baby needs continuous monitoring of saturation, heart rate, blood pressure, and respiratory rate, with the infusion titrated to the minimum dose that maintains adequate saturations and perfusion. The team must be ready to intubate at any point because apnoea can occur even after days of stable infusion. [10]
Prostaglandin E1 (alprostadil) in suspected duct-dependent CHD
The long-term outlook depends on the lesion, the timing of intervention, and whether there was a period of severe acidosis or shock before the duct was reopened. Lesions that can be repaired in a single operation, such as transposition of the great arteries, carry an excellent prognosis. Lesions that require staged palliation into a single-ventricle Fontan pathway, such as hypoplastic left heart syndrome, have improved dramatically but carry a lifelong burden of surgeries, complications, and surveillance. Neurodevelopmental outcome is a concern in all survivors of critical CHD, and structured follow-up is the standard of care. [3]
Specific Subtypes & Scenarios
The scenarios below are the ones that present, collapse, and are misdiagnosed, so they are the highest-yield material for the bedside and the written paper. [9]
Critical coarctation of the aorta is the classic missed duct-dependent systemic lesion. The baby is pink and well at discharge because the open duct is perfusing the lower body, and the femoral pulses may still be felt. On day three or four the duct closes, the lower body loses its blood supply, and the baby returns in shock with absent femoral pulses, severe metabolic acidosis, and poor perfusion. The history of a well baby and the finding of absent femoral pulses or a blood pressure differential is the diagnosis. Start prostaglandin immediately. [3]
Hypoplastic left heart syndrome is the most severe of the duct-dependent systemic lesions and carries the most complex surgical pathway. The left side of the heart is underdeveloped, and the right ventricle must pump blood both to the lungs and, through the ductus, to the body. When the duct closes the baby collapses with shock, poor perfusion, and deepening acidosis. The management is prostaglandin E1, staged palliation (Norwood, Glenn, Fontan), and lifelong cardiac follow-up. [3]
Pulmonary atresia with intact ventricular septum is the lesion that presents with deep cyanosis from birth or the first day, because the only route to the lungs is through the ductus. There is typically no murmur, and the saturation is very low and does not respond to oxygen. The prostaglandin infusion is life-saving, and the definitive management depends on the right ventricular morphology and coronary supply, ranging from catheter-based pulmonary valvotomy to staged single-ventricle palliation. [3]
Severe tetralogy of Fallot with a severely obstructive right ventricular outflow tract can present as a duct-dependent pulmonary lesion when the obstruction is critical. These babies may have spells of deep cyanosis — Tet spells — that worsen as the infundibular obstruction increases with crying or feeding. The prostaglandin keeps the duct open as a source of pulmonary blood flow until definitive surgical repair. [3]
Complications & Pitfalls
The complications divide into those of the untreated disease and those of its treatment. Untreated or late-treated duct-dependent CHD brings severe metabolic acidosis, multi-organ failure, cardiogenic shock, and death. The brain is particularly vulnerable: a prolonged period of low cardiac output before the duct is reopened can cause hypoxic-ischaemic brain injury, and neurodevelopmental impairment is a recognised long-term outcome in survivors of critical CHD who had preoperative shock. [3]
The lethal pitfall is the missed diagnosis. The baby who is sent home with critical coarctation or hypoplastic left heart syndrome and returns on day four in extremis is the preventable death, and the countermeasure is pulse oximetry screening and a low threshold to start prostaglandin E1 in any neonate with unexplained cyanosis, shock, or collapse. One infusion of prostaglandin is harmless if you are wrong and life-saving if you are right. [9]
Mortality of duct-dependent CHD if the duct closes without intervention
critical
Without prostaglandin E1, duct-dependent CHD is uniformly fatal within hours to days of duct closure. Pulse oximetry screening has reduced but not eliminated late presentations. The single most important determinant of survival is the time from duct closure to prostaglandin infusion.
The treatment pitfalls are concrete. Waiting for the echocardiogram before starting prostaglandin costs lives. Failing to anticipate apnoea and sending a baby on prostaglandin without intubation equipment risks an airway disaster during transport. Attributing fever to the prostaglandin infusion and missing sepsis is a recurring error. And stopping the prostaglandin before surgery, or failing to secure a reliable infusion pump, can cause sudden reclosure of the duct and collapse. [10]
Prognosis & Disposition
With early recognition, prostaglandin E1, and timely surgery, the outlook for most duct-dependent lesions has transformed. Lesions that can be repaired in a single operation — such as transposition of the great arteries after the arterial switch operation — now carry survival into the high nineties percent. Lesions requiring staged single-ventricle palliation — such as hypoplastic left heart syndrome — carry a more guarded but substantially improved prognosis compared to the pre-surgical era. The single biggest modifiable factor is whether the diagnosis is made before the duct closes. [3]
Neurodevelopmental outcome is a concern for all survivors of critical CHD. The combination of preoperative instability, cardiopulmonary bypass, and deep hypothermic circulatory arrest means that a proportion of children have cognitive, motor, and behavioural challenges that emerge over years. Structured neurodevelopmental follow-up, early intervention services, and school support are part of the standard of care for these children. [3]
Disposition is lifelong. The child needs a named cardiologist, a primary-care partner who understands the anatomy and the prostaglandin rules, a MedicAlert, a school care plan, and a structured transition to adult congenital cardiology. The family needs to understand the signs of decompensation — worsening cyanosis, poor feeding, lethargy — and have a clear pathway back to the cardiac centre. The time spent building the family's understanding and the transition plan is the highest-yield long-term clinical act. [3]
Special Populations
The neonate with an antenatally diagnosed duct-dependent lesion is the ideal scenario. Antenatal diagnosis allows planned delivery at or near a cardiac centre, prostaglandin to be started immediately after birth, and transfer to be avoided entirely. When the diagnosis is made in utero, the baby never goes through the collapse, and outcomes are better. The challenge is that antenatal detection rates vary widely by lesion, operator, and geography, and the left-sided obstructions that are most often missed postnatally are also the ones most often missed antenatally. [9]
The neonate in a rural or remote setting faces the combined challenge of distance from a cardiac centre and a narrower clinical team. The retrieval pathway — starting prostaglandin locally, intubating for transport, and organising a dedicated neonatal transport team — is the lifeline. In Australia and Aotearoa New Zealand, the regional neonatal retrieval networks coordinate these transfers, and the standard is that any neonate started on prostaglandin is discussed with a cardiac centre within the hour and transferred as soon as the team arrives. [8]
In Australia and Aotearoa New Zealand, pulse oximetry screening is embedded in routine newborn examination, and the regional paediatric cardiology networks provide 24-hour telephone consultation and retrieval. The challenge is equity: remote and Indigenous communities, and migrant and refugee families, carry a higher burden of late presentation because of distance, language, and access barriers. Culturally safe education for families about the warning signs, and reliable access to prostaglandin and retrieval, are determinants of survival in these communities. [8]
The child with a known chromosomal syndrome — 22q11 deletion, Turner, Noonan, or Down syndrome — carries a higher risk of critical CHD and should have a low threshold for cardiac evaluation. The 22q11 deletion in particular is associated with conotruncal lesions such as interrupted aortic arch and tetralogy of Fallot, both of which can be duct-dependent. A dysmorphic neonate with a heart lesion should have a chromosomal microarray. [3]
Evidence, Guidelines & Regional Differences
The evidence base for pulse oximetry screening rests on a series of landmark studies. The 2009 Swedish prospective screening study by de-Wahl Granelli and colleagues showed that adding pulse oximetry to routine newborn examination nearly halved the rate of undetected duct-dependent lesions and reduced the proportion of babies discharged home with a critical cardiac lesion. The 2011 PulseOx study by Ewer and colleagues in the Lancet provided the test-accuracy data in over 20,000 UK babies, and the 2012 meta-analysis by Thangaratinam pooled 13 studies and nearly 230,000 babies to give the definitive sensitivity and specificity estimates. [5] [6] [7]
The American Heart Association and American Academy of Pediatrics joint scientific statement, led by Mahle in 2009, codified the screening protocol and the thresholds, and the 2011 Pediatrics paper by Kemper translated that into implementation guidance for hospitals and health systems. The screening protocol — right hand and either foot, after 24 hours, fail at 90 percent or below, repeat at 95 percent or below — is now the global standard and has been adopted by health systems across North America, Europe, and Australasia. [4] [8]
The prostaglandin evidence is older but unchanged. Lewis and colleagues established the side-effect profile in their 1981 Circulation paper, and the dosing, the side effects, and the need for airway vigilance have been confirmed in four decades of subsequent use. The 2026 review in the American Journal of Emergency Medicine by Grabinski and colleagues synthesises the current approach to cyanotic critical congenital heart disease for the emergency physician, and reinforces the principle that prostaglandin is started on suspicion. [10] [11]
The remaining controversy is the optimal screening strategy. Pulse oximetry alone misses about a quarter of critical CHD, and the combination of antenatal ultrasound, postnatal examination, and pulse oximetry still leaves a small residual of missed lesions. The debate is whether to add a third modality — such as routine echocardiography in selected populations — and how to close the equity gap for remote and underserved communities where the screening infrastructure is weakest. [7]
Exam Pearls
One-sentence answer: the approach to a neonate with suspected duct-dependent CHD
A neonate who collapses on day two to five with cyanosis unresponsive to oxygen, or with shock and weak pulses, has duct-dependent CHD until proven otherwise: start prostaglandin E1 at 0.01 to 0.05 micrograms per kilogram per minute immediately, check the glucose and blood gas, intubate if apnoeic, cover sepsis with cultures and antibiotics, confirm the anatomy with echocardiography, and transfer to a cardiac centre for definitive surgery.
The two pathways
- Duct-dependent pulmonary: right-sided obstruction → presents with CYANOSIS (pulmonary atresia, critical PS, severe TOF, tricuspid atresia, Ebstein)
- Duct-dependent systemic: left-sided obstruction → presents with SHOCK (HLHS, critical coarctation, interrupted aortic arch, critical AS, mitral atresia)
- Both collapse on day 2 to 5 when the duct closes; both are saved by prostaglandin E1
- The echo confirms the lesion but must NOT delay the prostaglandin
The resuscitation
- Start prostaglandin E1 (alprostadil) 0.01 µg/kg/min IV, titrate to 0.05 µg/kg/min
- Intubate if apnoeic or before transport (apnoea in ~10% on PGE1)
- Cover sepsis — it mimics and coexists with duct-dependent CHD
- Check glucose, venous gas, 4-limb BP, CXR; echo when stable
Screening and detection
- Pulse oximetry: right hand + foot, after 24 hours; fail if SpO₂ <90% or differential >3%
- Pulse oximetry alone detects ~75% of critical CHD; combined screening reaches >90%
- Hyperoxia test: PaO₂ <100 mmHg on 100% O₂ suggests fixed right-to-left shunt
- Most missed lesion is critical coarctation — check femoral pulses in every neonate
Frequently misremembered facts, stated correctly: the most commonly missed duct-dependent lesion is critical coarctation, not HLHS, because the baby looks pink on day one; the hyperoxia test uses a PaO2 threshold of 100 millimetres of mercury, not 50; prostaglandin E1 causes apnoea, fever, and hypotension, and the pyrexia can mask sepsis; and the femoral pulses can still be felt on day one even in critical coarctation because the duct is still open. [9] [10]
The lesion-to-presentation pairings are the fastest route to a bedside answer and the highest-yield material for a written or oral question: absent femoral pulses with shock in a day-three neonate is critical coarctation; deep cyanosis from birth with no murmur is pulmonary atresia; a well baby who collapses on day four with hepatomegaly is HLHS; and cyanosis that does not correct with 100 percent oxygen is cardiac until proven otherwise. [3] [11]
References
- [1]Hoffman JI; Kaplan S The incidence of congenital heart disease. J Am Coll Cardiol, 2002.PMID 12084585
- [2]van der Linde D; Konings EE; Slager MA; et al Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol, 2011.PMID 22078432
- [3]Silberbach M; Hannon D Presentation of congenital heart disease in the neonate and young infant. Pediatr Rev, 2007.PMID 17400823
- [4]Mahle WT; Newburger JW; Matherne GP; et al Role of pulse oximetry in examining newborns for congenital heart disease: a scientific statement from the AHA and AAP. Pediatrics, 2009.PMID 19581259
- [5]de-Wahl Granelli A; Wennergren M; Sandberg K; et al Impact of pulse oximetry screening on the detection of duct dependent congenital heart disease: a Swedish prospective screening study in 39,821 newborns. BMJ, 2009.PMID 19131383
- [6]Ewer AK; Middleton LJ; Furmston AT; et al Pulse oximetry screening for congenital heart defects in newborn infants (PulseOx): a test accuracy study. Lancet, 2011.PMID 21820732
- [7]Thangaratinam S; Brown K; Zamora J; et al Pulse oximetry screening for critical congenital heart defects in asymptomatic newborn babies: a systematic review and meta-analysis. Lancet, 2012.PMID 22554860
- [8]Kemper AR; Mahle WT; Martin GR; et al Strategies for implementing screening for critical congenital heart disease. Pediatrics, 2011.PMID 21987707
- [9]Wren C; Reinhardt Z; Khawaja K Twenty-year trends in diagnosis of life-threatening neonatal cardiovascular malformations. Arch Dis Child Fetal Neonatal Ed, 2008.PMID 17556383
- [10]Lewis AB; Freed MD; Heymann MA; Roach A; Rudolph AM Side effects of therapy with prostaglandin E1 in infants with critical congenital heart disease. Circulation, 1981.PMID 7285304
- [11]Grabinski Z; Bhat R; Ahmed R; et al High risk and low incidence diseases: Cyanotic critical congenital heart disease. Am J Emerg Med, 2026.PMID 41237673