Paeds · cardiology
Ebstein anomaly and tricuspid valve disease
Also known as Ebstein anomaly · Ebstein's anomaly · Ebstein malformation · Apical tricuspid displacement · Tricuspid valve dysplasia · Congenital tricuspid regurgitation · Tricuspid atresia · Cone repair
Fellowship guide to Ebstein anomaly and tricuspid valve disease in children: the apically displaced tricuspid valve from failed delamination, the hugely dilated right atrium swallowing a small functional right ventricle, the neonate who is ductal-dependent when functional pulmonary atresia closes off forward flow, the Carpentier grades and Great Ormond Street score that predict outcome, the cone reconstruction that rebuilds a competent valve, and the lifelong surveillance for tricuspid regurgitation, arrhythmia and sudden death.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single idea that holds the whole topic together is that Ebstein anomaly is a disease of tricuspid-valve leaflet formation: because the septal and posterior leaflets are tethered down into the right ventricle, the valve cannot close, the right atrium balloons, and everything that follows — the tricuspid regurgitation, the cyanosis, the arrhythmia, the surgical strategy — flows from that one developmental error. Grasp the anatomy and the rest is mechanism. [1] [4]
This page covers the recognition of Ebstein anomaly from fetus to adult, the Carpentier grades and Great Ormond Street score that predict outcome, the neonatal resuscitation of the ductal-dependent severe form, the cone reconstruction and the single-ventricle Starnes pathway for the most severe cases, the arrhythmia burden, and the lifelong follow-up that no Ebstein patient ever leaves. It links to the ductal-dependent congenital heart disease and neonatal cyanosis leaves for the broader differential. [2] [5]
Overview & Definition
Ebstein anomaly is defined by the apical displacement of the functional tricuspid-valve orifice toward the right ventricular apex, produced by failed delamination of the septal and posterior leaflets during cardiac development. The anterior leaflet is usually large and sail-like but tethered, and the effective coaptation point sits far below the true fibrous annulus. The right ventricle is partitioned into a thin, atrialised inflow portion that is electrically ventricular but haemodynamically part of the atrium, and a small, often poorly contracting functional outflow portion. [1] [4]
What makes the lesion behave as it does is the combination of a regurgitant valve and a small pumping chamber. Tricuspid regurgitation floods the right atrium, which dilates enormously and may hold the rightward and inferior interventricular septum flat against the left ventricle, impairing left-sided filling. A right-to-left shunt across a patent foramen ovale or atrial septal defect is present in most patients, producing cyanosis and a risk of paradoxical embolism. When the regurgitation is severe enough that the functional right ventricle cannot generate forward flow across the pulmonary valve, the neonate has functional pulmonary atresia and depends on the ductus arteriosus for pulmonary blood flow. [5] [2]
Tricuspid-valve disease in children beyond Ebstein includes tricuspid dysplasia (regurgitant valve leaflets without displacement), tricuspid atresia (absent valve and a hypoplastic right ventricle producing ductal-dependent cyanosis), and the rarer tricuspid stenosis of Noonan syndrome, carcinoid or rheumatic disease. Ebstein anomaly is the dominant congenital tricuspid lesion, and the one a general paediatrician must recognise at the bedside. [11] [7]
Classification
Grade the lesion by how far the valve is displaced and how much it obstructs the right ventricular outflow, because the grade predicts both the age of presentation and the surgical options. The figure splits Ebstein anomaly into the four Carpentier grades of increasing severity and places it within the wider spectrum of tricuspid-valve disease. [4] [8]

Carpentier A (mild)
- Minimal apical displacement; RV volume near-normal
- Often asymptomatic; found on incidental murmur or echo
- May never need surgery; lifelong surveillance only
- Best prognosis of the four grades
Carpentier B (moderate)
- Displaced leaflets but no RV outflow obstruction
- Variable tricuspid regurgitation; RV may dilate
- Presents in childhood or adolescence with fatigue
- Cone repair when symptomatic or RV dilates
Carpentier C–D (severe)
- Restricted anterior leaflet → RV outflow obstruction (C)
- Tricuspid sack: nearly all RV atrialised (D)
- Neonatal heart failure, cyanosis, ductal dependence
- Starnes single-ventricle pathway often required
The Great Ormond Street Echocardiography (GOSE) score grades neonatal severity and is the single best predictor of survival in the symptomatic neonate. It combines four echocardiographic features — the degree of tricuspid-regurgitant-jet displacement, the ratio of the right atrium plus atrialised right ventricle to the functional right ventricle, and the presence of right-ventricular outflow obstruction — into a score from one to four. A score of one or two carries a good prognosis with medical or surgical management; a score of three or four carries a high mortality and is the threshold at which single-ventricle palliation or even transplant is considered. [10] [3]
Epidemiology & Risk Factors
Ebstein anomaly is rare, accounting for less than one percent of all congenital heart defects, with an estimated incidence of roughly one in twenty thousand live births. Because mild cases may never be diagnosed, the true prevalence is probably higher, and adult-onset presentation after decades of asymptomatic disease is well described. The lesion affects both sexes roughly equally, though some series report a slight female predominance. [1] [7]
The one established environmental risk factor is first-trimester lithium exposure, which raises the relative risk several-fold, although the absolute risk remains low and modern practice does not routinely deny lithium to women with severe psychiatric illness. Maternal benzodiazepine use has also been implicated in some studies. Most cases are sporadic, but familial recurrence and association with left-heart lesions, myopathies and channelopathies point to a genetic contribution that is increasingly being mapped. [11] [4]
A clinically important association is Ebstein anomaly with left ventricular non-compaction, now recognised as a distinct cardiomyopathy-overlap phenotype that carries its own systolic-dysfunction and arrhythmia risk. MYH7 and other sarcomeric and cardiomyopathy gene variants recur in Ebstein cohorts, and syndromic forms occur with CHD7 (CHARGE) and other developmental gene defects. The practical implication is that a genetics referral and, where appropriate, a cardiomyopathy gene panel are part of the modern work-up, especially when there is a family history or extracardiac features. [11] [9]
Pathophysiology
To understand why an Ebstein baby is cyanosed, picture the right heart as a pump whose inlet valve has slipped down into the chamber it is meant to guard. The septal and posterior leaflets never separated from the ventricular wall, so they cannot coapt; the anterior leaflet is a large, sail-like sheet but tethered and ineffective. Every right ventricular systole therefore ejects a large fraction of its stroke volume backwards into the right atrium rather than forwards into the pulmonary artery. [5] [2]
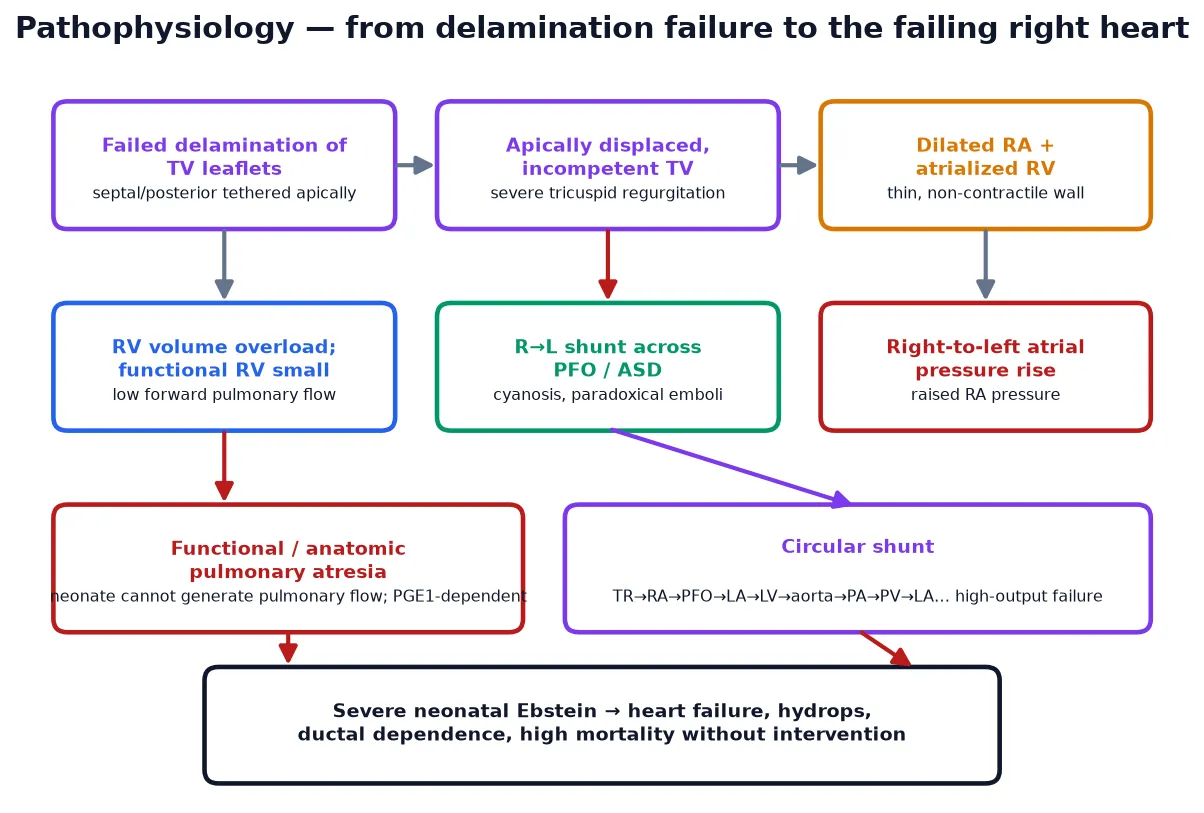
The dilated right atrium and atrialised right ventricle swallow the bulk of right-heart volume, while the functional right ventricle — the part that actually contracts — is small and thin. When the right atrial pressure rises above the left, the foramen ovale or an atrial septal defect opens right-to-left, shunting desaturated blood into the systemic circulation and producing cyanosis. The dilated right heart also flattens the interventricular septum against the left ventricle, reducing left-ventricular filling and output, which is why even the left heart underperforms in severe disease. [1] [2]
In the most severe neonatal form, the right ventricle is so dysfunctional and the tricuspid regurgitation so torrential that no blood crosses the pulmonary valve at all — this is functional pulmonary atresia, where the valve is anatomically patent but functionally closed. The neonate is then entirely dependent on the ductus arteriosus for pulmonary blood flow, and closing the duct produces catastrophic hypoxaemia. Some severely affected neonates develop a circular shunt in which blood recirculates between the right atrium, atrial defect, left heart, aorta and ductus without ever being effectively oxygenated, a state that is rapidly fatal unless interrupted. [10] [12]
Clinical Presentation
The presentation spans the whole of life and depends entirely on severity. The severe neonatal form declares itself within hours to days of birth with deep cyanosis, a hyperdynamic precordium from the volume-overloaded right heart, a loud tricuspid regurgitant murmur, hepatomegaly, and signs of low output. The chest X-ray shows a hugely enlarged heart — the so-called wall-to-wall heart — with diminished pulmonary vascular markings because forward pulmonary flow is poor. This is the ductal-dependent Ebstein neonate who needs prostaglandin E1 and urgent cardiology input. [5] [10]
The milder infant or child presents with less dramatic but characteristic features: exercise intolerance and fatigue disproportionate to the examination, a systolic tricuspid regurgitant murmur that may be loud and scratching, a widely split second heart sound, and sometimes the tricuspid-opening sound and triple rhythm that hint at the diagnosis. Cyanosis may be absent, intermittent, or present only on exertion, and an incidental murmur or an abnormal chest X-ray may be the first clue. An adolescent or adult may present for the first time with palpitations, pre-syncope or sustained arrhythmia, or with a paradoxical embolic event. [7] [4]
The right-to-left atrial shunt that accompanies most Ebstein patients carries two specific risks that candidates must name. Paradoxical embolism can send venous thrombus into the cerebral circulation, causing stroke, and because the shunt bypasses the pulmonary capillary filter, a cerebral abscess can follow even minor bacteraemia. This is why air filters on intravenous lines, counselling against decompression diving, and a low threshold for endocarditis prophylaxis in high-risk dental and surgical procedures are part of routine Ebstein care. [1] [7]
Differential Diagnosis
The differential in the cyanosed neonate is the broader ductal-dependent right-heart and pulmonary-vascular differential, and the key is to recognise that the huge heart of Ebstein distinguishes it from the small, oligaemic lung fields of pulmonary atresia with intact ventricular septum or the normal-sized heart of transposition. Pulmonary disease causes cyanosis with a normal or large heart and usually responds to oxygen, whereas Ebstein does not improve with oxygen because the right-to-left shunt persists. [5] [2]
Points to Ebstein
- Massively enlarged heart on chest X-ray ('wall-to-wall')
- Tricuspid regurgitant murmur; multiple heart sounds
- Cyanosis that does not improve with oxygen
- Apically displaced tricuspid valve on echocardiography
- GOSE score grades neonatal severity
Points to a mimic
- Pulmonary atresia + intact septum: small heart, oligaemic lungs
- Transposition: cyanosis, narrow mediastinum, improves with PGE1
- Persistent pulmonary hypertension: structural heart normal
- Tricuspid atresia: hypoplastic RV, no TR murmur, ductal-dependent
- Neonatal sepsis or pneumonia: response to oxygen, inflammatory markers
In the older child or adolescent, the differential of a systolic murmur with right-heart enlargement includes pulmonary hypertension with secondary tricuspid regurgitation, an atrial septal defect with right-heart volume overload, and a primary arrhythmia presenting before the structural lesion is recognised. An echocardiogram that specifically images the tricuspid-valve leaflet insertion resolves the question, and any patient with a suspected accessory pathway should have the valve imaged to exclude Ebstein. [7] [4]
Tricuspid dysplasia — a regurgitant valve with malformed but normally positioned leaflets — is the closest mimic and is distinguished from Ebstein only by the absence of apical displacement on echocardiography. The distinction matters because the two share the haemodynamic consequences but may differ in surgical approach and in the associated genetic syndromes. Tricuspid atresia is the other key tricuspid lesion in the neonatal cyanosis differential, but its absent tricuspid-valve tissue, hypoplastic right ventricle and obligatory atrial-level right-to-left shunt make it clinically and echocardiographically distinct. [11] [5]
Clinical & Bedside Assessment
The focused bedside assessment of a neonate with suspected Ebstein rests on recognising the huge, underperfused, cyanosed heart that does not respond to oxygen. Inspect for central cyanosis, a hyperactive precordium and a visibly enlarged abdomen from hepatomegaly. Auscultate for the loud pansystolic tricuspid regurgitant murmur at the lower left sternal edge, the widely split second sound, and the additional sounds — a tricuspid-opening sound and a third heart sound — that the dilated right heart produces. [5] [4]
Measure pre- and post-ductal saturations, because the right-to-left atrial shunt produces systemic desaturation that may differ between the upper and lower body if the shunt includes the duct. Check the blood gas for metabolic acidosis, which reflects the low systemic output of the severe form, and feel the femoral pulses, which may be weak in the low-output state. In the older child, assess exercise tolerance formally, listen for the murmur, and examine for the stigmata of right-heart failure — raised jugular venous pressure, hepatomegaly and peripheral oedema. [1] [2]
A chest X-ray at the bedside often gives the diagnosis before the echocardiogram: the massively enlarged heart with a right atrial bulge and diminished pulmonary vascular markings is the classic Ebstein picture. An electrocardiogram adds supporting evidence — right atrial enlargement with tall P waves, a right bundle branch block, and in a fifth of patients the delta wave and short PR interval of pre-excitation that flags the accessory pathway. Finding pre-excitation on the ECG of a child with a murmur should trigger an echocardiogram to look for Ebstein. [9] [7]
Investigations
Echocardiography is the definitive investigation and establishes the diagnosis, grades the severity and defines the surgical anatomy in a single study. The key finding is the apical displacement of the tricuspid septal leaflet hinge point by more than eight millimetres per square metre of body surface area above the mitral annulus, with tethering of the septal and posterior leaflets and a sail-like anterior leaflet. Doppler quantifies the tricuspid regurgitation, estimates the pulmonary pressure, and identifies the right-to-left atrial shunt. [2] [6]
Echocardiography
- First-line and definitive — displacement, leaflet morphology, TR
- GOSE score grades neonatal severity and predicts survival
- Assesses RV size, function and outflow obstruction
- Identifies ASD/PFO, pulmonary atresia, associated lesions
ECG and rhythm
- Right atrial enlargement (tall P waves), right bundle branch block
- Pre-excitation / delta wave in ~20% — accessory pathway
- Holter for occult atrial arrhythmia in older children
- Electrophysiology study if accessory pathway or SVT
Adjunct imaging & genetics
- Cardiac MRI: RV volumes and function, when echo windows poor
- Chest X-ray: 'wall-to-wall' heart, oligoaemic lung fields
- Genetic referral: MYH7, cardiomyopathy panel, LVNC overlap
- Fetal echo: GOSE score, hydrops, prenatal planning
Cardiac magnetic resonance imaging adds precision in the older child and adult, where it quantifies right-ventricular volumes and ejection fraction more accurately than echocardiography and helps decide the timing of surgery. In the fetus, echocardiography defines the tricuspid displacement, grades the GOSE score, and looks for hydrops, which is the gravest prenatal sign and the trigger for considering prenatal therapy. [7] [10]
Genetic evaluation is now part of the comprehensive work-up, particularly when there is a family history, extracardic features, or coexistent left-ventricular non-compaction. A cardiomyopathy gene panel including MYH7 captures a meaningful fraction of familial and syndromic cases, and identifying a pathogenic variant informs cascade screening of relatives. The systematic evidence for the Ebstein–LVNC overlap supports this integrated approach. [11] [9]
Management — Resuscitation
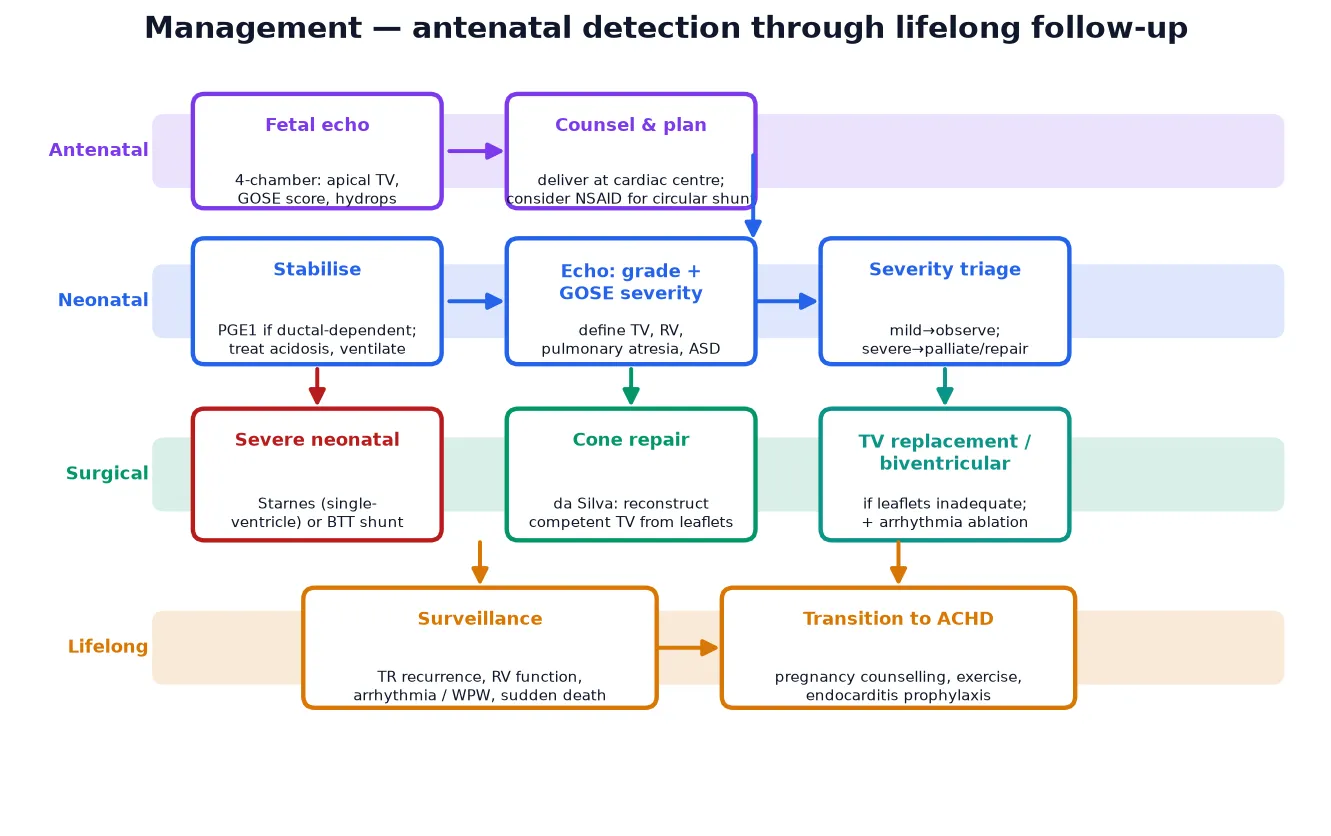
The resuscitation of the severely cyanosed Ebstein neonate rests on keeping the duct open and supporting the failing right heart while the pulmonary vascular resistance falls. A neonate with functional pulmonary atresia has no effective forward pulmonary flow except through the ductus arteriosus, so prostaglandin E1 is started immediately to maintain ductal patency until the natural fall in pulmonary vascular resistance allows the functional right ventricle to generate forward flow. [5] [3]
Prostaglandin E1 (alprostadil) is given at 0.01 to 0.05 micrograms per kilogram per minute, titrated to the oxygenation response, with the usual vigilance for apnoea, hypotension and fever. The team must be ready to intubate, and a baby on prostaglandin E1 being retrieved to a cardiac centre must travel intubated or with airway expertise, because apnoea in transit is a recognised preventable death. The aim is to bridge the neonate through the first days of life while the pulmonary vascular resistance falls and the right ventricle, if it has enough functional capacity, begins to sustain forward flow. [3] [12]
Supportive measures run alongside the ductal therapy. Correct the metabolic acidosis with fluid and inotropic support, maintain normothermia and glucose, and avoid anything that raises pulmonary vascular resistance, such as acidosis or hypoxia. In the rare fetus with a circular shunt, prenatal non-steroidal anti-inflammatory therapy to constrict or close the duct has been reported to interrupt the shunt and reverse hydrops, an evolving intervention offered only in specialist fetal centres. Once the neonate is stabilised, the GOSE score and the echocardiographic anatomy drive the decision between biventricular repair and single-ventricle palliation. [3] [10]
Management — Definitive & Stepwise
Definitive surgical management depends on the severity and the age at presentation. For the symptomatic infant, child or adult with moderate-to-severe Ebstein anomaly and a functional right ventricle, the procedure of choice is the cone reconstruction, in which the surgeon mobilises the available leaflet tissue — chiefly the large anterior leaflet — and rotates and plicates it into a cone-shaped competent tricuspid valve that coapts at the true annulus. The atrialised right ventricle is plicated, the right atrium is reduced, and any atrial septal defect is closed. [8] [6]
The cone reconstruction, introduced by da Silva in 2007, transformed Ebstein surgery by producing a valve that more closely resembles a normal tricuspid valve than earlier repairs, and medium-term follow-up shows durable freedom from reoperation and good right-ventricular remodelling in most patients. The comparison of cone against conventional (monocusp or annular) repair favours the cone for long-term valve competence, and it is now the standard against which other techniques are measured. Concomitant arrhythmia surgery — accessory-pathway division or a right-atrial maze — is performed at the same sitting when indicated. [8] [6]
For the most severe neonatal Ebstein, where the functional right ventricle is too small to sustain a biventricular circulation, the modified Starnes procedure is the accepted palliation: the tricuspid valve is partially closed to eliminate the regurgitation, an atrial septectomy secures right-to-left decompression, and a systemic-to-pulmonary shunt provides pulmonary blood flow, committing the child to a single-ventricle Fontan pathway. Long-term follow-up of Starnes survivors shows acceptable single-ventricle outcomes, and the procedure is the bridge that keeps the most severe neonates alive to reach staged palliation. Tricuspid-valve replacement is reserved for the patient whose leaflets cannot be reconstructed, and heart transplantation is the final option for end-stage disease. [12] [3]
Specific Subtypes & Scenarios
The severe neonatal Ebstein with functional pulmonary atresia is the scenario the acute exam tests. A cyanosed neonate with a wall-to-wall heart, a loud tricuspid regurgitant murmur and metabolic acidosis is ductal-dependent until proven otherwise. The management is prostaglandin E1, ventilatory and inotropic support, echocardiographic grading by the GOSE score, and triage to either biventricular repair or the Starnes single-ventricle pathway. The pitfall is attributing the cyanosis to pulmonary disease and waiting for oxygen to work, which delays the ductal therapy that sustains life. [10] [12]
Ebstein with Wolff-Parkinson-White and atrial arrhythmia is the scenario that tests the arrhythmia link. Roughly one in five Ebstein patients has an accessory pathway, and atrial tachyarrhythmias — atrioventricular re-entrant tachycardia, atrial flutter and atrial fibrillation — are the dominant late problem and the leading cause of sudden death. The long-term multicentre paediatric study of arrhythmias in Ebstein documents this burden, and the modern approach is electrophysiological study with accessory-pathway ablation, either before surgery or concomitantly. Any Ebstein patient with new palpitations or syncope needs urgent rhythm evaluation. [9] [7]
The adolescent or adult with previously undiagnosed Ebstein is the transition scenario. A patient who was mildly affected in childhood may present for the first time in adolescence or adulthood with fatigue, exercise intolerance, new arrhythmia, a paradoxical embolic event, or right-heart failure. The cone repair is effective across the age range, and the decision to operate is driven by symptoms, right-ventricular size and function, and the arrhythmia burden. The transition to adult congenital heart disease services must address pregnancy counselling, exercise guidance, endocarditis prophylaxis and ongoing arrhythmia surveillance. [7] [2]
Ebstein with left ventricular non-compaction is the scenario that tests the genetic and cardiomyopathy link. The systematic review of this overlap phenotype documents that coexistent non-compaction carries an additional systolic-dysfunction and arrhythmia risk, and that sarcomeric gene variants — particularly MYH7 — recur in these families. The practical implication is that any Ebstein patient with left-ventricular dysfunction, a family history, or extracardic features earns a cardiomyopathy gene panel and a genetics referral, and relatives are offered screening. [11] [9]
Complications & Pitfalls
The untreated severe Ebstein neonate dies of low-output heart failure, hypoxaemia and acidosis, often with hydrops in the fetal counterpart. The dominant late complications in survivors and in the milder cohort are progressive tricuspid regurgitation with right-heart dilation and failure, atrial and ventricular arrhythmia with the risk of sudden death, and paradoxical embolism through the persistent atrial shunt. Each of these is a target of lifelong surveillance. [4] [9]
After surgical repair, the chief complications are recurrent tricuspid regurgitation, residual right-ventricular dysfunction, and late arrhythmia. Cone reconstruction has reduced the reoperation rate compared with earlier techniques, but a subset of patients still require reoperation for valve failure, and tricuspid-valve replacement is the salvage option. The atrial arrhythmia burden may persist or re-emerge after surgery, which is why concomitant arrhythmia surgery and ongoing rhythm surveillance are integral to the long-term plan. [6] [7]
The avoidable pitfalls cluster around three failures. Missing the ductal dependence in the severe neonate delays prostaglandin E1 and is fatal. Ignoring the arrhythmia in the older child or adult allows an accessory pathway to cause sudden death before surgical or ablation intervention. Forgetting the right-to-left shunt leads to paradoxical embolism from unfiltered intravenous lines or decompression diving, both preventable with an air filter and simple counselling. Each pitfall is rooted in forgetting one defining feature of the disease. [3] [11]
Prognosis & Disposition
Prognosis is driven by severity at presentation. The neonatal GOSE score is the strongest single predictor: a score of one or two carries a good prognosis with medical or surgical management, while a score of three or four carries a high mortality and is the threshold for single-ventricle palliation or consideration of transplant. Prenatal diagnosis with hydrops carries the worst prognosis of all, and early referral to a fetal cardiac centre is essential because prenatal therapy may be the only intervention that changes outcome. [10] [3]
For the milder cohort who reach childhood or adulthood, the prognosis with modern cone repair is good, with durable valve competence and acceptable survival in medium-term follow-up. The lifelong battleground is the late complications — recurrent regurgitation, right-ventricular failure, and arrhythmia — which is why no Ebstein patient is ever discharged from cardiac surveillance. The comparison of cone against conventional repair supports cone as the standard for long-term valve function. [6] [8]
Disposition is to a tertiary paediatric cardiac centre for the acute presentation and any surgical intervention, then to a structured transition pathway into adult congenital heart disease services. The general paediatrician's role is to recognise the lesion — in the fetus, the neonate, or the older child — to start the ductal therapy in the acute case, to coordinate retrieval, and then to champion the lifelong surveillance and the transition that the patient needs across the decades. [1] [7]
Special Populations
The fetus with Ebstein anomaly and hydrops is the highest-risk cohort. Prenatal echocardiography grades the GOSE score and looks for hydrops, and where the score is three or four or hydrops is present, the prognosis is grave. Prenatal non-steroidal anti-inflammatory therapy to close a circular shunt and reverse hydrops is an evolving intervention offered in specialist fetal centres, and early referral is essential because the window for prenatal therapy is narrow. [3] [10]
The adolescent or adult with previously undiagnosed Ebstein carries the accumulated burden of decades of tricuspid regurgitation, right-heart dilation and arrhythmia. The transition to adult congenital heart disease services must address pregnancy counselling — pregnancy is generally well tolerated in milder disease but carries risk with significant right-heart dysfunction or arrhythmia — exercise guidance, endocarditis prophylaxis, and the ongoing rhythm surveillance that sudden-death prevention requires. [7] [2]
Patients with the Ebstein–LVNC overlap phenotype carry the combined risk of a regurgitant right-heart valve and a cardiomyopathic left ventricle. They need integrated cardiology and genetics input, a cardiomyopathy gene panel, and family screening, because the sarcomeric variants that underlie the overlap can be inherited and may declare in relatives. Remote and Indigenous families face the logistics of prolonged separation from home and community during the surgical admission, so a clear retrieval and repatriation pathway, telehealth follow-up and cultural support are essential to equitable care. [11] [9]
Evidence, Guidelines & Regional Differences
The evidence base for Ebstein anomaly has matured into lifespan reviews, surgical comparisons and a 2024 consensus guideline. The fetus-to-adult literature review by Ramcharan and colleagues (2022) sets out the contemporary pathway for patient care across the age range, and the multidisciplinary imaging-and-therapy review by Pasqualin and colleagues (2024) covers the modern approach. The AATS 2024 expert consensus by Konstantinov and colleagues is the current guideline for neonatal and infant management. [1] [2]
The surgical evidence rests on da Silva's original cone-reconstruction report (2007), which established the technique, and the cone-versus-conventional comparison by Burri and colleagues (2020), which documents the medium-term superiority of the cone for valve competence. The Starnes single-ventricle outcomes report by Kumar and colleagues (2016) anchors the palliative pathway for the most severe neonates, the systematic review of Ebstein with left-ventricular non-compaction by Thareja and colleagues (2022) documents the cardiomyopathy overlap, and the long-term multicentre paediatric arrhythmia study by Delhaas and colleagues (2010) anchors the arrhythmia burden. The neonatal-outcome study by Yu and colleagues (2013) and the beyond-childhood narrative review by Neumann and colleagues (2021) complete the lifespan evidence. [8] [6]
Regional practice differences are modest because the surgical and resuscitation principles are internationally adopted and the AATS 2024 consensus provides a shared standard, but access to paediatric cardiac surgery, neonatal retrieval, fetal cardiac services and prostaglandin E1 in remote areas varies. In Australia and New Zealand, suspected Ebstein anomaly is referred to a tertiary paediatric cardiac centre, with retrieval services trained to start prostaglandin E1 in transit and fetal cardiac services centralised in the major cities. The main controversies are the timing of surgery in the asymptomatic dilating right ventricle, the threshold for single-ventricle versus biventricular repair in the borderline neonate, and the role of prenatal therapy for the hydropic fetus. [3] [7]
Exam Pearls
Hold one sentence for the viva: a cyanosed neonate with a wall-to-wall heart, a loud tricuspid regurgitant murmur and functional pulmonary atresia has severe Ebstein anomaly and needs prostaglandin E1 to keep the duct open until the pulmonary vascular resistance falls. [5] [10]
State the frequently tested facts correctly. The defining lesion is failed delamination of the tricuspid septal and posterior leaflets, producing apical displacement of the true valve orifice. Severity is graded by the Carpentier types A to D and, in the neonate, by the GOSE score, where a score of three or four predicts high mortality. The neonate with functional pulmonary atresia is ductal-dependent and gets prostaglandin E1 at 0.01 to 0.05 micrograms per kilogram per minute. The cone reconstruction, introduced by da Silva, is the procedure of choice for the repairable valve; the modified Starnes is the single-ventricle palliation for the most severe neonate. [8] [12]
The high-yield pairings: a neonate with a huge heart and cyanosis that does not improve with oxygen is Ebstein until the echo excludes it; an Ebstein patient with palpitations has an accessory pathway until the ECG and electrophysiology exclude it; an Ebstein patient with left-ventricular dysfunction may have the LVNC overlap phenotype and needs a cardiomyopathy gene panel; a hydropic fetus with Ebstein is GOSE three or four and warrants urgent fetal-cardiac referral. These pairings do most of the diagnostic and management work in the short and long cases. [9] [11]
References
- [1]Ramcharan TKW; Goff DA; Greenleaf CE; et al Ebstein's Anomaly: From Fetus to Adult-Literature Review and Pathway for Patient Care. Pediatr Cardiol, 2022.PMID 35460366
- [2]Pasqualin G; Boccellino A; Chessa M; et al Ebstein's anomaly in children and adults: multidisciplinary insights into imaging and therapy. Heart, 2024.PMID 37487694
- [3]Konstantinov IE; Chai P; Bacha E; et al The American Association for Thoracic Surgery (AATS) 2024 expert consensus document: Management of neonates and infants with Ebstein anomaly. J Thorac Cardiovasc Surg, 2024.PMID 38685467
- [4]Holst KA; Connolly HM; Dearani JA Ebstein's Anomaly. Methodist Debakey Cardiovasc J, 2019.PMID 31384377
- [5]Galea J; Ellul S; Schembri A; et al Ebstein anomaly: a review. Neonatal Netw, 2014.PMID 25161135
- [6]Burri M; Mrad Agua K; Cleuziou J; et al Cone versus conventional repair for Ebstein's anomaly. J Thorac Cardiovasc Surg, 2020.PMID 32711971
- [7]Neumann S; Rüffer A; Sachweh J; et al Narrative review of Ebstein's anomaly beyond childhood: Imaging, surgery, and future perspectives. Cardiovasc Diagn Ther, 2021.PMID 35070800
- [8]da Silva JP; Baumgratz JF; da Fonseca L; et al The cone reconstruction of the tricuspid valve in Ebstein's anomaly. The operation: early and midterm results. J Thorac Cardiovasc Surg, 2007.PMID 17198815
- [9]Delhaas T; Sarvaas GJ; Rijlaarsdam ME; et al A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol, 2010.PMID 19937010
- [10]Yu JJ; Yun TJ; Won HS; et al Outcome of neonates with Ebstein's anomaly in the current era. Pediatr Cardiol, 2013.PMID 23494543
- [11]Thareja SK; Frommelt MA; Lincoln J; et al A Systematic Review of Ebstein's Anomaly with Left Ventricular Noncompaction. J Cardiovasc Dev Dis, 2022.PMID 35448091
- [12]Kumar SR; Kung G; Noh N; et al Single-Ventricle Outcomes After Neonatal Palliation of Severe Ebstein Anomaly With Modified Starnes Procedure. Circulation, 2016.PMID 27777295