Paeds · cardiology
Hypoplastic left heart syndrome
Also known as Hypoplastic left heart syndrome · HLHS · Single-ventricle physiology of the left heart · Aortic and mitral atresia · Norwood sequence · Univentricular heart of the left-heart type
Fellowship guide to hypoplastic left heart syndrome: the most severe ductal-dependent systemic-outflow obstruction, presenting as the day-two-to-four neonatal collapse with weak pulses and acidosis; the prostaglandin-E1-first resuscitation before the echo; the echocardiographic anatomy of atretic mitral and aortic valves with a hypoplastic ascending aorta and a right ventricle doing all the work; and the three-stage single-ventricle palliation — Norwood, Glenn, Fontan — whose SVR-trial evidence, interstage mortality and neurodevelopmental burden every candidate must own.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single idea that organises this topic is that the right ventricle does all the work, and the ductus arteriosus is the lifeline: while the duct is open the baby survives, and when it closes the baby dies unless prostaglandin E1 reopens it. Every clinical feature, every resuscitation step and every surgical stage flows from that mechanism. [2] [3]
This page covers the recognition and neonatal stabilisation of HLHS, the echocardiographic anatomy, the three-stage palliation and the SVR-trial evidence behind the shunt choice, the interstage mortality that is the dominant modifiable risk, the neurodevelopmental burden, and the lifelong follow-up every Fontan survivor needs. It links to the ductal-dependent congenital heart disease leaf for the broader differential and to the neonatal cyanosis leaf for the saturation-based approach. [1] [2]
Overview & Definition
HLHS is not a single defect but a spectrum of underdevelopment of the left heart. The mitral and aortic valves are atretic or critically stenotic, the left ventricle is a slit-like or absent cavity that cannot generate systemic pressure, and the ascending aorta and arch are hypoplastic — sometimes thread-like — because they never carried meaningful forward flow in fetal life. The right ventricle is structurally normal and becomes the systemic ventricle, pumping the whole circulation. [2]
What makes HLHS lethal without intervention is its absolute ductal dependence. The right ventricle ejects into the pulmonary artery, and in HLHS the only route for that blood to reach the body is retrograde through the ductus arteriosus into the descending aorta, and from there back up the hypoplastic arch to the head, neck and coronary arteries. While the duct is patent in fetal life and the early hours after birth, this parallel circulation sustains the child. The moment the duct constricts, systemic output collapses. [3]
HLHS sits at the severe end of the ductal-dependent congenital heart disease family, alongside coarctation, interrupted aortic arch and critical aortic stenosis. The unifying principle for the general paediatrician is that any neonate who collapses after a period of wellbeing, with shock, acidosis and weak pulses, has a ductal-dependent lesion until proven otherwise and gets prostaglandin E1 before the echo. [2]
Classification
A candidate sorts HLHS along two axes that carry different weight in the viva: the valve anatomy, which predicts severity and surgical risk, and the management strategy, which frames the whole life course. The figure below splits the lesion by its mitral and aortic valve morphology and by the staged palliation that every patient enters. [2] [3]
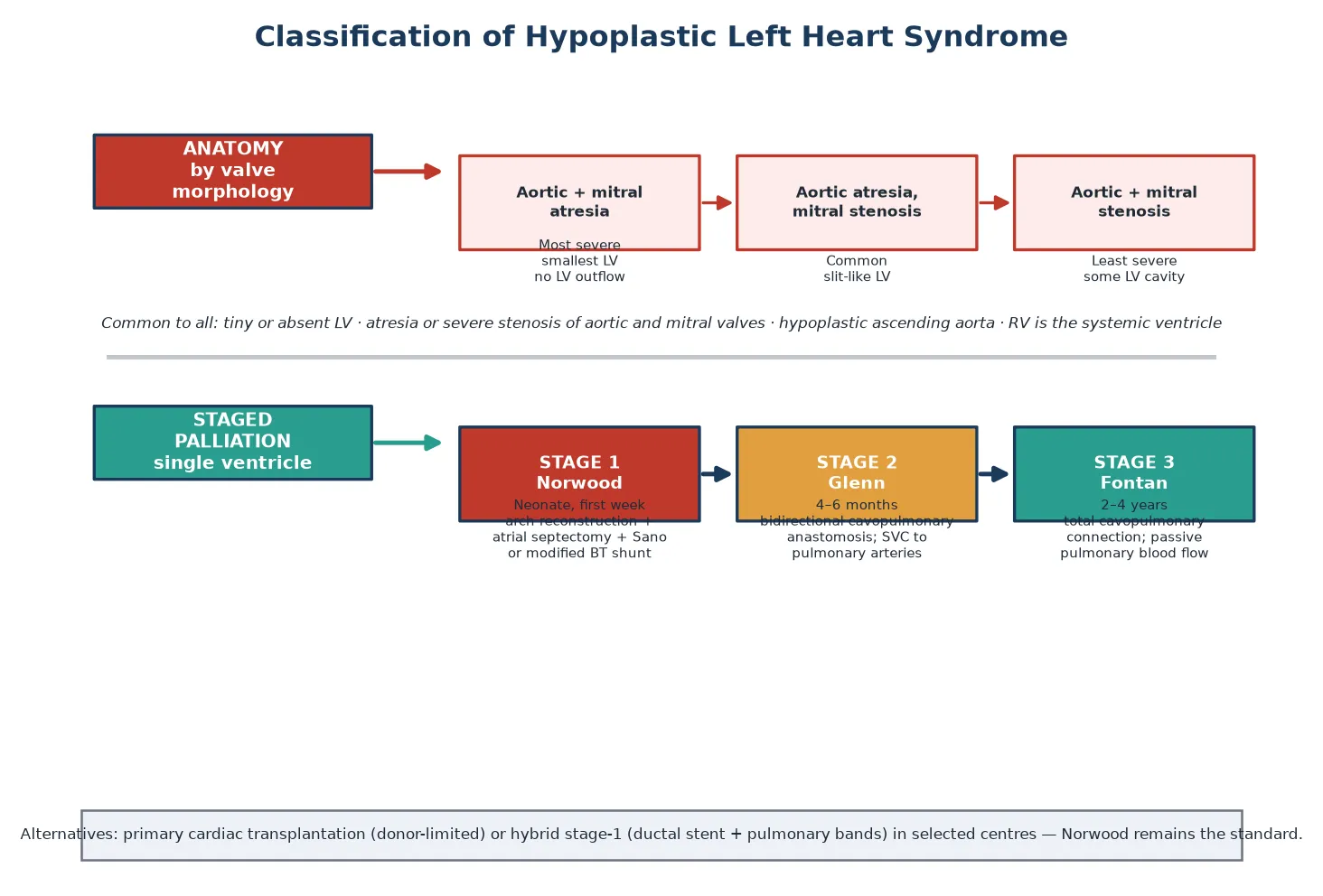
Aortic + mitral atresia
- Most severe subtype; smallest or absent LV cavity
- No left-ventricular outflow at all
- Ascending aorta often thread-like, perfused retrograde
- Highest early surgical risk
Aortic atresia, mitral stenosis
- Commonest subtype
- Small slit-like LV still present
- Some antegrade flow possible in fetal life
- Intermediate surgical risk
Aortic + mitral stenosis
- Least severe subtype
- Larger LV cavity with critically reduced inflow and outflow
- Borderline with critical aortic stenosis
- May be considered for biventricular repair in expert hands
The management strategy is the second axis. Standard care is staged single-ventricle palliation, in which the right ventricle is deliberately kept as the systemic pump and the venous return is routed directly to the lungs in stages. The alternatives are primary cardiac transplantation, which is donor-limited and leaves the child on the waitlist during the high-risk neonatal period, and the hybrid stage-one procedure — a ductal stent plus pulmonary artery bands — used in selected centres for high-risk infants or as a bridge. The Norwood procedure remains the standard first stage. [2] [1]
Epidemiology & Risk Factors
HLHS accounts for roughly two to three percent of all congenital heart defects and occurs in about two to three infants per ten thousand live births, making it one of the commoner severe lesions a general paediatrician will encounter in the neonatal period. It is the most common cause of cardiac death in the first week of life when undiagnosed. There is a male predominance of roughly two to one. [2]
The associations a fellowship candidate must name are the genetic syndromes and extracardiac anomalies that change the surgical candidacy and the long-term plan. Turner syndrome, Noonan syndrome, Holt-Oram syndrome and Jacobsen syndrome are recognised associations, and chromosomal abnormalities including trisomy 13 and 18 coexist in a substantial minority. A right aortic arch, anomalous pulmonary venous drainage and other left-sided obstructive lesions in the same family point to a monogenic aetiology with recurrence risk. [2] [3]
The reason these associations matter is that they change counselling and management. A syndromic infant has higher perioperative mortality and a heavier neurodevelopmental burden, which shapes the goals-of-care discussion before the first operation. A family with a previous left-sided obstructive lesion earns recurrence-risk counselling and dedicated fetal echocardiography in subsequent pregnancies. Recognising the genetic context is part of recognising the whole child. [3] [10]
Pathophysiology
To understand why an HLHS baby collapses, picture the fetal circulation as a system that routes right-ventricular output through the pulmonary artery and across the ductus arteriosus into the descending aorta. Because the left ventricle is underdeveloped and the mitral and aortic valves are atretic, essentially no blood traverses the left heart in fetal life. The right ventricle carries the combined cardiac output, and the duct is the conduit to the systemic circulation. The fetus grows normally because this parallel circuit is perfectly adequate in utero. [2] [3]
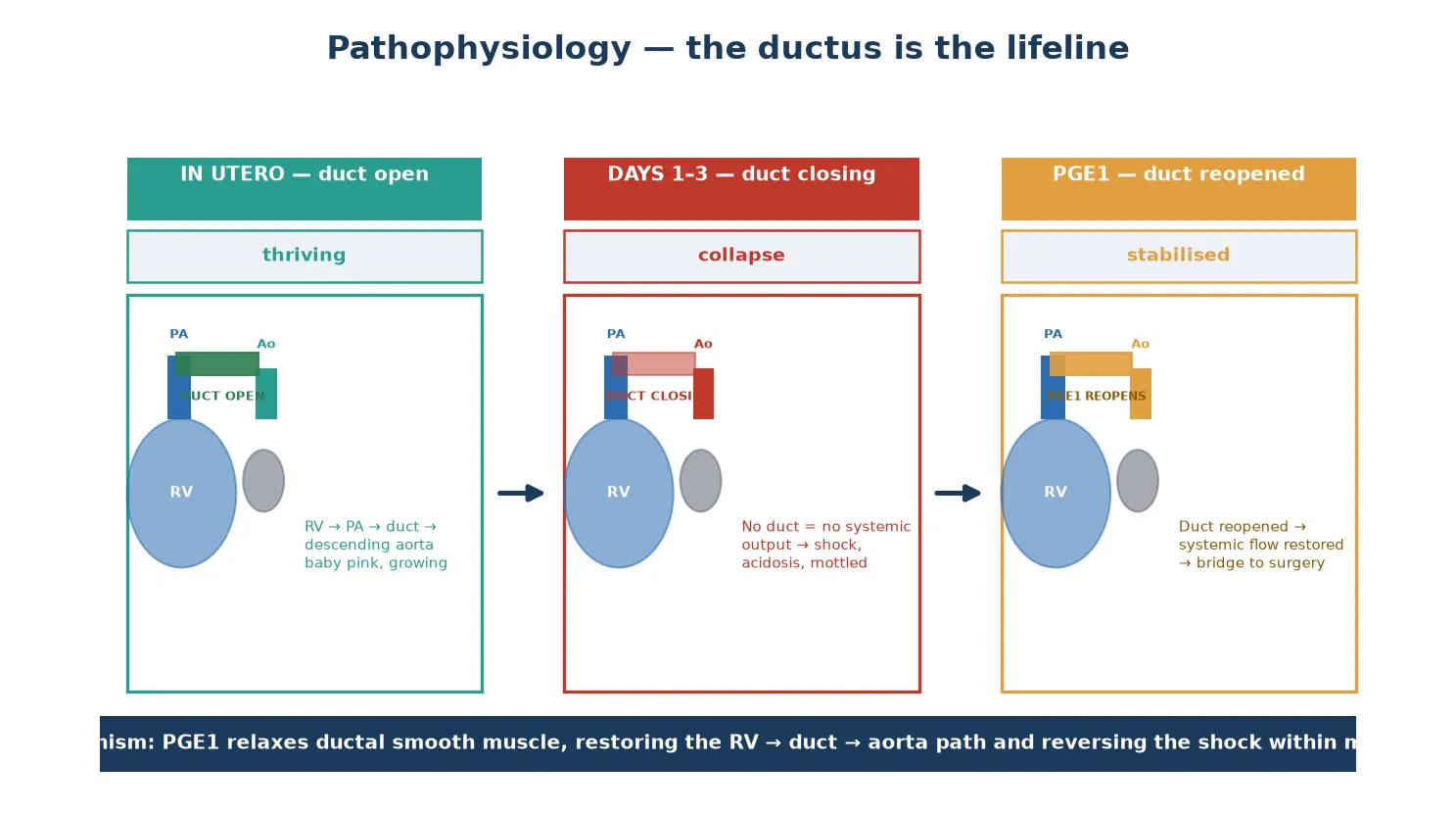
The collapse is driven by a biological clock, not a random event. As oxygen tension rises after birth, the ductal smooth muscle constricts over hours to days, progressively narrowing the only systemic route. In a normal baby this is harmless; in an HLHS baby it chokes systemic output entirely. The child becomes mottled and oliguric, develops progressive metabolic acidosis, and proceeds to cardiovascular arrest. The window between the first signs and arrest can be short, which is why empirical prostaglandin E1 is mandatory in any collapsing neonate. [2]
The atrial septum is the second critical structure. Pulmonary venous blood returning to the left atrium must cross the atrial septum to reach the right ventricle, the only pumping chamber. A restrictive atrial septum traps pulmonary venous return, causing severe pulmonary oedema and profound hypoxaemia — a preoperative emergency that demands an urgent balloon atrial septostomy or atrial stent rather than escalation of oxygen. Prostaglandin E1 relaxes ductal smooth muscle, reopening the duct within minutes and restoring the right-ventricle-to-duct-to-aorta path that buys time for diagnosis and surgery. [3]
Clinical Presentation
A neonate with HLHS presents in one of two ways: the dramatic neonatal collapse, or the antenatally detected baby managed from birth. The dramatic presentation is the classic exam scenario. A baby who was feeding and thriving for the first two to four days of life becomes tachypnoeic, grey and mottled, with poor feeding and progressive lethargy, then shock and metabolic acidosis as the duct constricts. The single most important bedside sign is uniformly weak pulses — brachial and femoral together — in a shocked neonate, because the right ventricle can no longer sustain systemic pressure through the narrowing duct. [2] [3]
The antenatally detected baby presents differently and increasingly commonly. A fetal echocardiogram at the twenty-week anomaly scan or a targeted cardiac scan identifies the atretic mitral and aortic valves, the hypoplastic ascending aorta and the reversed flow in the duct, and the family is counselled, the delivery is planned at a cardiac centre, and prostaglandin E1 is started at the first clinical sign. Prenatal diagnosis does not eliminate collapse but it shortens the time to definitive treatment and improves the interstage outcome. [3]
Cyanosis in HLHS may be mild at first, because systemic and pulmonary blood are both derived from the right ventricle and mixing occurs. As pulmonary blood flow rises or as the systemic output falls, the cyanosis deepens and the shock picture dominates. Unlike coarctation, where the upper body stays pink and the legs go blue, HLHS produces generalised hypoperfusion because all systemic flow depends on the struggling right ventricle. [2] [3]
Differential Diagnosis
The differential turns on separating HLHS from the other ductal-dependent lesions and from the medical causes of neonatal collapse. The key bedside discriminator is the pulse pattern. In HLHS all pulses are weak because systemic output is failing; in coarctation and interrupted aortic arch the upper-limb pulses remain strong while the femorals vanish. This single distinction does much of the work at the bedside. [2] [3]
Points to HLHS
- Uniformly weak brachial and femoral pulses in a shocked neonate
- Grey, mottled generalised hypoperfusion rather than differential cyanosis
- Single second heart sound; often no murmur
- Metabolic acidosis with rising lactate over hours
- Echo: atretic mitral/aortic valves, hypoplastic ascending aorta
Points to a mimic
- Coarctation/IAA: strong brachial, absent femoral, differential cyanosis
- Critical aortic stenosis: harsh murmur, pulmonary oedema, weak all pulses
- Transposition: profound differential cyanosis, healthy myocardium
- Septic shock: fever, normal saturations, no structural lesion on echo
- Cardiomyopathy/myocarditis: gallop, hepatomegaly, globally poor function
Sepsis is the most common misattribution. A collapsing HLHS baby looks like late-onset sepsis, and the temptation is to pursue a septic workup and antibiotics while the duct continues to close. The disciplines that prevent this error are to palpate all four limb pulses in every shocked neonate, to check pre- and post-ductal saturations, and to start prostaglandin E1 empirically whenever a ductal-dependent lesion is possible — the antibiotics can be given alongside, but the ductal-dependent lesion is the time-critical diagnosis. [2] [3]
Clinical & Bedside Assessment
The focused assessment of a neonate with suspected HLHS rests on three bedside manoeuvres: palpate all four limb pulses, measure pre- and post-ductal saturations, and assess the perfusion. In HLHS the pulses are uniformly weak rather than differentially absent, which distinguishes it from arch obstruction. The pre- and post-ductal saturations are often both low, and a rising lactate or falling pH on the blood gas confirms the failing systemic output. [2]
Auscultation reveals a single second heart sound, because the aortic component is absent and only the pulmonary valve closes. A systolic murmur may be soft or absent, because there is no high-velocity flow across the atretic valves; its absence does not exclude HLHS and must not be reassuring. Hepatomegaly reflects the failing right ventricle, and a gallop rhythm may be present. The key teaching point is that an unremarkable auscultatory examination in a collapsing neonate is a ductal-dependent lesion, not reassurance. [3]
Examine for the syndromic stigmata that flag an associated condition. Short stature, webbed neck and a low posterior hairline suggest Turner syndrome. Dysmorphic features, a cleft palate and hypocalcaemia point to 22q11.2 deletion, which coexists in a minority and changes the airway and perioperative plan. A thorough dysmorphology examination at the bedside often predicts the genetic work-up and shapes the counselling before surgery. [2] [3]
Investigations
Echocardiography is the definitive investigation and almost always the only imaging needed before surgery. A complete study defines the mitral and aortic valve morphology (atresia versus stenosis), the size of the left ventricle, the calibre of the ascending aorta and arch, the patency and direction of ductal flow, the atrial septum (restrictive or not), the tricuspid valve competence, and the right-ventricular function. It also excludes anomalous pulmonary venous drainage, which is a surgical game-changer. [2] [3]
Echocardiography
- First-line and definitive; defines the whole anatomy
- Shows mitral/aortic atresia, LV size, arch calibre
- Assesses atrial septum, duct, tricuspid valve, RV function
- Excludes anomalous pulmonary venous drainage
Supporting tests
- Blood gas: metabolic acidosis with rising lactate in collapse
- Four-limb blood pressures: low and often equal
- Genetics: microarray, karyotype for associated syndromes
- Renal and liver function: end-organ injury from hypoperfusion
Adjuncts
- Chest X-ray: pulmonary oedema if atrial septum restrictive
- ECG: right-axis deviation, right-ventricular hypertrophy
- Brain imaging if concern for hypoxic-ischaemic injury
- Head ultrasound before any circulatory arrest
The blood gas is the bedside measure of severity. A metabolic acidosis with a rising lactate reflects the degree of systemic hypoperfusion and should improve as prostaglandin E1 restores ductal flow. Renal and hepatic function may show acute injury from the low-output state, and these trends guide the timing of surgery. Genetic testing with chromosomal microarray is part of the first work-up because syndromic status changes surgical candidacy and counselling. [2]
Management — Resuscitation
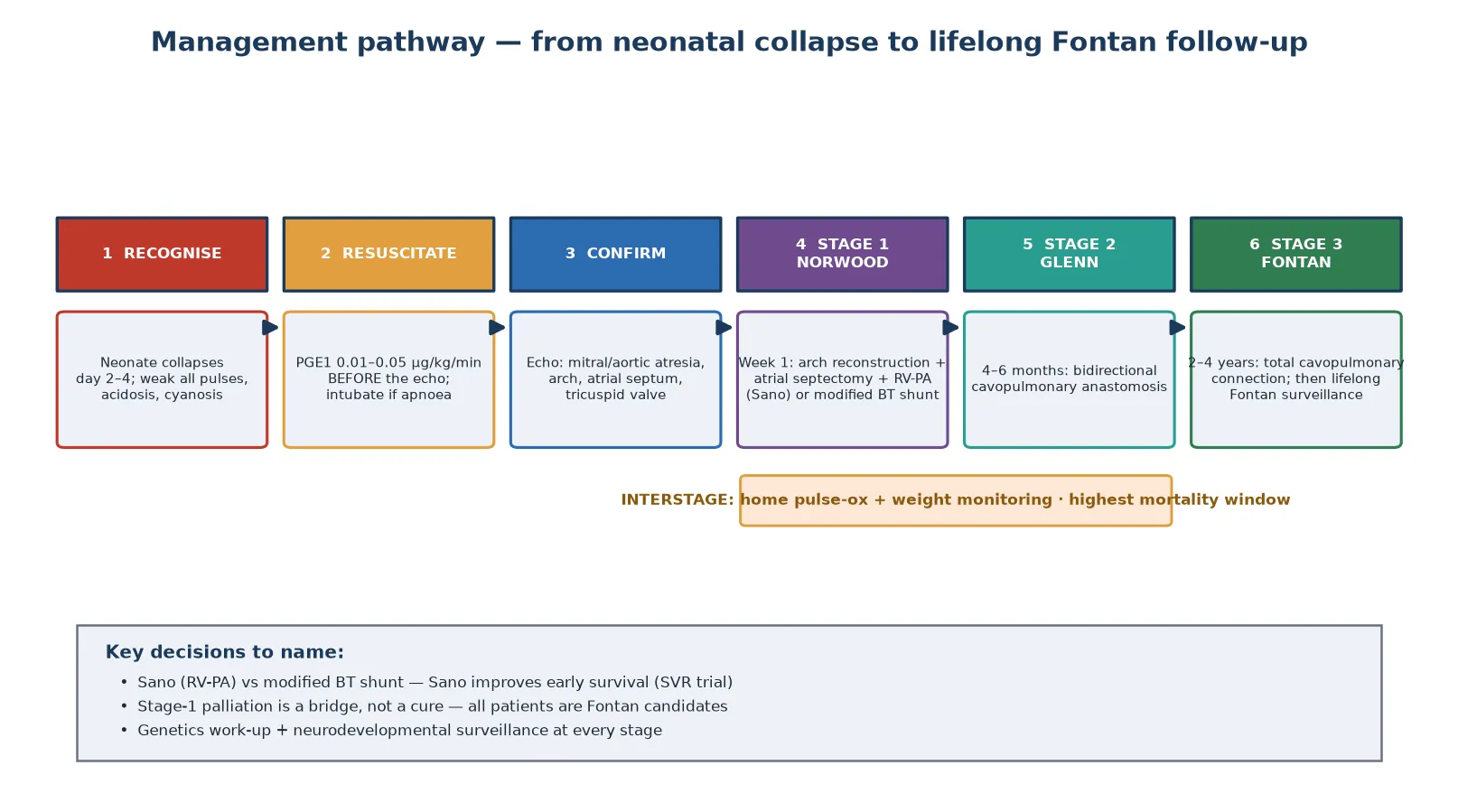
The resuscitation of a neonate with HLHS rests on one drug and one principle: prostaglandin E1 to reopen the duct, started before the diagnosis is confirmed. A collapsing neonate with weak pulses gets the infusion empirically. The downside of an unnecessary dose — apnoea, hypotension, fever — is manageable; the downside of delay is death. This is the single highest-yield fact in the topic. [2] [3]
Prostaglandin E1 (alprostadil) is started at 0.01 to 0.05 micrograms per kilogram per minute and titrated to the clinical response. The duct usually reopens within thirty minutes to a few hours, restoring systemic flow, improving perfusion and correcting the metabolic acidosis. Apnoea is the most important side effect: a significant minority of infants on PGE1 needs intubation, so the team must be ready to ventilate. A baby on PGE1 being retrieved to a cardiac centre must travel intubated or with skilled airway support. [2]
Balancing the pulmonary and systemic blood flow is the second resuscitation discipline. After the duct is open, the right ventricle ejects into both circuits, and the ratio of pulmonary to systemic flow — the Qp to Qs ratio — determines perfusion. Hyperventilation and high oxygen drive pulmonary vasodilation, steal flow from the systemic circuit and worsen the shock, so the infant is ventilated to a normal carbon dioxide target on the lowest oxygen that maintains acceptable saturations. A restrictive atrial septum demands an urgent balloon atrial septostomy or stent. [3]
Management — Definitive & Stepwise
Definitive treatment is staged single-ventricle palliation, in which the right ventricle is deliberately maintained as the systemic pump and the venous return is routed to the lungs in three operations spread across the first years of life. The pathway is Norwood, then Glenn, then Fontan. The figure below maps the six steps from neonatal recognition to lifelong follow-up. [1] [2]
The stage-one Norwood operation is performed in the first week of life and is the highest-risk procedure in the pathway. It has three components: reconstruction of the hypoplastic aortic arch from the pulmonary artery to create a systemic outflow, an atrial septectomy to allow pulmonary venous return to reach the right ventricle, and a source of pulmonary blood flow — either a right-ventricle-to-pulmonary-artery conduit (the Sano modification) or a modified Blalock-Taussig shunt. The Single Ventricle Reconstruction trial established that the Sano conduit improves transplant-free survival through the first year compared with the modified Blalock-Taussig shunt, though with a higher rate of later interventions. [1] [9]
The stage-two bidirectional Glenn operation is performed at four to six months, once the pulmonary vascular resistance has fallen. The superior vena cava is connected directly to the pulmonary arteries, so that half the venous return flows passively to the lungs and off-loads the right ventricle. The stage-three Fontan operation at two to four years completes the total cavopulmonary connection, directing all systemic venous return to the lungs and leaving the right ventricle as a pure systemic pump. After the Fontan, the patient enters lifelong surveillance for the complications of the single-ventricle circulation. [2] [8]
Specific Subtypes & Scenarios
The neonate with a restrictive atrial septum is the scenario that tests the candidate on the second emergency in HLHS. A restrictive atrial septum traps pulmonary venous return behind the left atrium, causing profound hypoxaemia and pulmonary oedema that does not respond to oxygen. The intervention is an urgent balloon atrial septostomy or, where that fails, an atrial stent placed in the catheter laboratory, performed alongside the prostaglandin infusion. Missing a restrictive atrial septum condemns the child to intractable hypoxaemia before surgery. [3]
The antenatally diagnosed infant is the scenario that tests prenatal counselling and planned management. When HLHS is identified on fetal echocardiography, the family is counselled about the three-stage pathway and its burden, the delivery is planned at or near a cardiac centre, and prostaglandin E1 is started at the first clinical sign. Prenatal diagnosis shortens the time to definitive treatment and is associated with improved interstage outcomes, though it does not eliminate the risk of collapse. [3] [4]
The interstage infant is the scenario that tests knowledge of the highest-mortality window. Between the Norwood discharge and the Glenn, the child depends on a shunt for pulmonary blood flow and is vulnerable to sudden circulatory collapse from shunt thrombosis, respiratory illness or feeding failure. Home monitoring with daily pulse oximetry and weight, rapid access to the cardiac centre, and parental education on the warning signs are the interventions that reduce interstage death. Any fever, tachypnoea or colour change in an interstage infant is presumed circulatory failure until proven otherwise. [4] [7]
The Fontan survivor in late follow-up is the scenario the long case tests. A patient years after the Fontan may present with arrhythmia, protein-losing enteropathy, thromboembolism, Fontan failure with ascites and peripheral oedema, or progressive exercise intolerance. The candidate must frame the Fontan as a functional single-ventricle circulation with predictable late complications, not a cure, and must name the lifelong surveillance that every survivor needs. [8] [9]
Complications & Pitfalls
The untreated HLHS lesion is lethal: without intervention, the ductal closure causes progressive shock, hypoxic-ischaemic injury to the brain and other organs, and death, typically within the first weeks of life. The single biggest modifiable prognostic factor is the speed of recognition and ductal reopening with prostaglandin E1, which is why empirical treatment before the echo is non-negotiable. [2] [3]
The dominant modifiable complication of the palliated pathway is interstage mortality. The period between Norwood discharge and the Glenn carries the highest risk of death in the whole life course, driven by shunt thrombosis, circulatory failure from intercurrent illness, and feeding and growth failure. The SVR trial quantified this risk and showed that home surveillance programmes — daily pulse oximetry and weight, parental education, and rapid access — reduce interstage death. A validated risk score identifies the highest-risk infants for closer monitoring. [4] [5]
The palliated survivor faces a defined set of late complications. Arrhythmias, especially atrial tachycardias after the Fontan, are common and may herald Fontan failure. Tricuspid regurgitation worsens as the systemic right ventricle dilates. Protein-losing enteropathy, plastic bronchitis and thromboembolism are the recognised Fontan-specific morbidities. The neurodevelopmental burden is substantial and persists into school age and adolescence. The treatment also carries its own pitfalls: PGE1 apnoea in transit, missing a restrictive atrial septum, and underestimating an interstage deterioration are each preventable harms rooted in forgetting the mechanism. [12] [9]
Prognosis & Disposition
With timely prostaglandin stabilisation and expert staged palliation, the outlook for HLHS has transformed from universally fatal to a chronic, survivable condition. Roughly seven to eight in ten infants now survive to and through the Fontan in modern cohorts, supported by the SVR-trial evidence, improved surgical technique and structured interstage programmes. Prenatal diagnosis and centre expertise further improve the outcome. [1] [9]
The long-term battleground is the late morbidity rather than early survival alone. Fontan survivors face a rising burden of arrhythmia, heart failure, reoperation and thromboembolism across the decades, and some ultimately require transplantation. The neurodevelopmental outcome is the other persistent concern: a substantial proportion of survivors have cognitive, attention and executive-function deficits that declare in school age and shape their educational and psychosocial trajectory. Long-term follow-up of SVR-trial participants documents this burden well into childhood. [8] [9]
Disposition is to a tertiary paediatric cardiac centre for the staged palliation, then to a structured transition pathway into adult congenital heart disease services. The general paediatrician's role is to recognise the lesion early, start prostaglandin E1, coordinate the retrieval, champion the interstage home monitoring, and then sustain the lifelong follow-up and neurodevelopmental surveillance the patient needs across the transition to adult care. [2] [10]
Special Populations
Neonates with prenatal diagnosis increasingly present as a managed rather than an emergency cohort. When HLHS is identified antenatally, the delivery is planned at a cardiac centre, the team is prepared, and prostaglandin E1 is started at the first sign. This shifts the outcome by eliminating the delay between collapse and treatment, and it allows the family to make an informed decision about the three-stage pathway before birth. [3] [4]
Infants with genetic syndromes or extracardiac anomalies carry a higher perioperative mortality and a heavier neurodevelopmental burden than isolated HLHS. Syndromic status changes surgical candidacy, intensifies the perioperative monitoring, and shapes the goals-of-care counselling. The candidate must name the common associations — Turner, Noonan, Holt-Oram, Jacobsen and chromosomal anomalies — and explain how they alter the plan. [2] [10]
Remote and Indigenous families face the logistics of prolonged separation from home and community during the surgical admissions and interstage period. A clear retrieval and repatriation pathway, telehealth follow-up for the interstage monitoring, cultural support, and accommodation near the cardiac centre are essential to equitable care. The general paediatrician often coordinates this wrap-around support across the geographic and cultural distance. [4] [7]
Evidence, Guidelines & Regional Differences
The evidence base for HLHS is anchored by the Single Ventricle Reconstruction trial, the largest randomised study in paediatric cardiac surgery. The original SVR trial (Ohye and colleagues, 2010) compared the right-ventricle-to-pulmonary-artery Sano conduit with the modified Blalock-Taussig shunt and showed better transplant-free survival with the Sano conduit, at the cost of more reinterventions. The six-year follow-up (Newburger and colleagues, 2018) and the longitudinal follow-up (Goldberg and colleagues, 2023) confirmed the shunt-type association and documented the long-term interventions and outcomes. [1] [8] [9]
The interstage evidence defines the modifiable risk. The multicentre SVR interstage analysis (Ghanayem and colleagues, 2012) quantified interstage mortality and its predictors, the home-monitoring programme data (Rudd and colleagues, 2020) supported surveillance with pulse oximetry and weight, and the validated risk score (Ahmed and colleagues, 2020) and interstage-death characteristics (Ahmed and colleagues, 2021) identified the highest-risk infants. The neurodevelopmental outcomes are anchored by the six-year neurodevelopmental study (Sananes and colleagues, 2021) and the school-age neuropsychological cohort (Bergemann and colleagues, 2015). The arrhythmia burden is documented in the Pediatric Heart Network analysis (Oster and colleagues, 2017). [4] [6] [5] [10]
Regional practice differences are modest because the surgical and resuscitation principles are internationally adopted, but access to paediatric cardiac surgery, neonatal retrieval and interstage support varies. In Australia and New Zealand, suspected or confirmed HLHS is referred to a tertiary paediatric cardiac centre, with retrieval services trained to start prostaglandin E1 in transit and intubate prophylactically. The main controversies are the Sano-versus-Blalock-Taussig shunt choice, the role of the hybrid stage-one in high-risk infants, the intensity of neurodevelopmental surveillance, and the timing and criteria for transplantation in Fontan failure. [2] [9]
Exam Pearls
Hold one sentence for the viva: a neonate who was well then collapses on day two to four with uniformly weak pulses, mottled skin and metabolic acidosis has a ductal-dependent lesion, and the treatment is prostaglandin E1 started before the echocardiogram confirms the diagnosis. [2] [3]
State the frequently tested facts correctly. The classic age of collapse is day two to four, at ductal closure. The bedside sign is uniformly weak pulses — all four limbs, not just femoral — which distinguishes HLHS from coarctation. Prostaglandin E1 at 0.01 to 0.05 micrograms per kilogram per minute reopens the duct. The single second heart sound reflects the absent aortic component. The Sano conduit (right-ventricle-to-pulmonary-artery) outperforms the modified Blalock-Taussig shunt on early survival, per the SVR trial. The interstage period is the highest-mortality window and is managed with home monitoring. [1] [4]
The high-yield pairings: a shocked neonate with uniformly weak pulses is HLHS until echo; a restrictive atrial septum with intractable hypoxaemia needs an urgent septostomy; an interstage infant with any deterioration is circulatory failure until proven otherwise; a Fontan survivor with arrhythmia or oedema has Fontan failure or its complications. These pairings do most of the diagnostic work in the short and long cases. [8] [9]
References
- [1]Ohye RG; Sleeper LA; Mahony L; et al Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med, 2010.PMID 20505177
- [2]Feinstein JA; Benson DW; Dubin AM; et al Hypoplastic left heart syndrome: current considerations and expectations. J Am Coll Cardiol, 2012.PMID 22192720
- [3]Rychik J Hypoplastic left heart syndrome: can we change the rules of the game? Circulation, 2014.PMID 25052402
- [4]Ghanayem NS; Allen KR; Tabbutt S; et al Interstage mortality after the Norwood procedure: Results of the multicenter Single Ventricle Reconstruction trial. J Thorac Cardiovasc Surg, 2012.PMID 22795436
- [5]Ahmed H; Anderson JB; Krawczeski PA; et al Development of a validated risk score for interstage death or transplant after stage I palliation for single-ventricle anatomy. J Thorac Cardiovasc Surg, 2020.PMID 31924360
- [6]Rudd NA; Ghanayem NS; Hill GD; et al Interstage Home Monitoring for Infants With Single Ventricle Heart Disease: Education and Management. J Am Heart Assoc, 2020.PMID 32777961
- [7]Ahmed H; Oster ME; Krawczeski PA; et al Characteristics of Interstage Death After Discharge from Stage I Palliation. Pediatr Cardiol, 2021.PMID 33948710
- [8]Newburger JW; Sleeper LA; Gaynor JW; et al Transplant-Free Survival and Interventions at 6 Years in the SVR Trial. Circulation, 2018.PMID 29437119
- [9]Goldberg CS; Trachtenberg FL; Krawczeski PA; et al Longitudinal Follow-Up of Children With HLHS and Association Between Norwood Shunt Type and Outcomes. Circulation, 2023.PMID 37795623
- [10]Sananes R; Goldberg CS; Bove EL; et al Six-Year Neurodevelopmental Outcomes for Children With Single-Ventricle Physiology. Pediatrics, 2021.PMID 33441486
- [11]Bergemann A; Hansen JH; Rotermann I; et al Neuropsychological performance of school-aged children after staged surgical palliation of hypoplastic left heart syndrome. Eur J Cardiothorac Surg, 2015.PMID 25100716
- [12]Oster ME; Chen S; Kugler JD; et al Development and impact of arrhythmias after the Norwood procedure: A report from the Pediatric Heart Network. J Thorac Cardiovasc Surg, 2017.PMID 27939495