Paeds · cardiology
Long-QT syndrome and channelopathies
Also known as LQTS · Long QT syndrome · Congenital long QT syndrome · Catecholaminergic polymorphic ventricular tachycardia · CPVT · Brugada syndrome · Inherited arrhythmia syndromes
Fellowship guide to long-QT syndrome and the inherited channelopathies in children: what a prolonged QTc means, the three cardinal genotypes (LQT1, LQT2, LQT3) and their trigger-specific biology, the Schwartz diagnostic score, the syncope-or-seizure child who needs a 12-lead ECG, beta-blocker-first management, left cardiac sympathetic denervation and ICD thresholds, and the related entities CPVT, Brugada syndrome and short-QT syndrome, with the AHA/HRS, ESC and CSANZ guideline positions.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the eight-year-old girl brought to the emergency department after collapsing during a school swimming carnival — she was pulled from the pool unconscious, briefly stiffened, and recovered fully within minutes. Her mother, who has fainted "all her life," wonders whether this was a fit. The triage label says "vasovagal or seizure — query epilepsy." That child carries the whole story of long-QT syndrome: an arrhythmic collapse triggered by exertion, mislabelled as a seizure or a faint, in a family with a heritable tendency to sudden death. One twelve-lead ECG, read for the QT interval, would have changed everything. [1] [2]
Long-QT syndrome is the commonest inherited cardiac arrhythmia syndrome, with a prevalence of about one in two thousand individuals. It is defined by prolongation of the corrected QT interval on the surface electrocardiogram, reflecting delayed repolarisation of the ventricular myocardium. The delayed repolarisation creates a vulnerable window in which early afterdepolarisations can arise, triggering the characteristic polymorphic ventricular tachycardia known as torsades de pointes, which may terminate spontaneously as a faint or progress to ventricular fibrillation and sudden cardiac death. [2] [10]
The clinical importance of LQTS rests on three facts. It is treatable: beta-blockers reduce events by roughly two-thirds and more aggressive therapy is available for the high-risk subset. It is under-diagnosed: syncope during exertion or emotion is routinely mislabelled as vasovagal or epileptic, and the first presentation in a family is sometimes the proband's sudden death. And it is heritable in an autosomal dominant pattern, so a diagnosis in one person carries implications for first-degree relatives who may be asymptomatic but at risk. [1] [6]
Classification
The most useful way to classify the channelopathies is by the ion-current abnormality and its genetic basis, because the genotype determines the trigger profile, the response to beta-blockers, and the threshold for aggressive therapy. Long-QT syndrome itself is subdivided by the affected ion channel into numbered subtypes (LQT1 through LQT17), of which the first three account for the vast majority of clinically relevant disease. [2] [12]
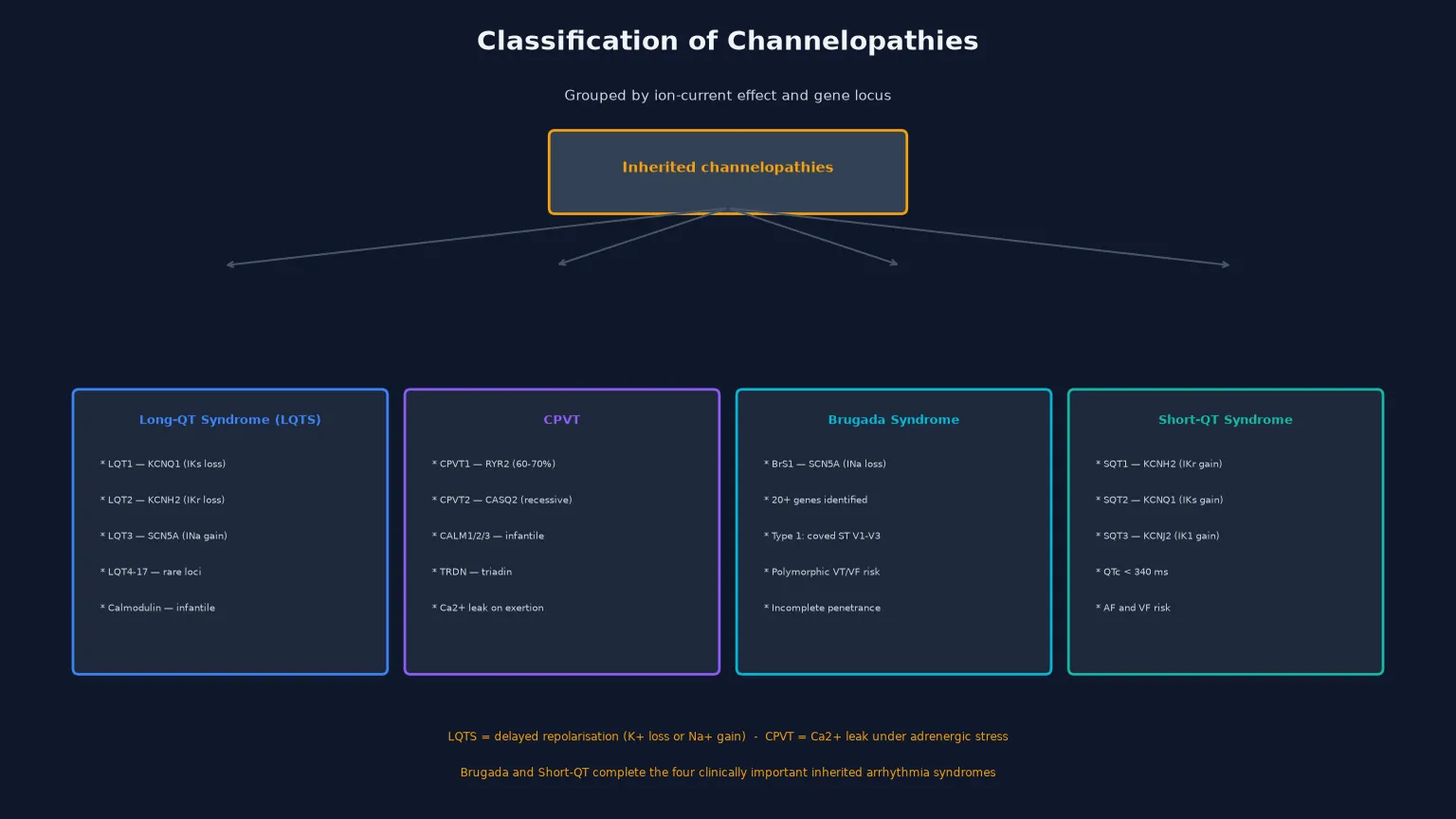
The three cardinal LQTS genotypes are LQT1 (KCNQ1, loss of the slow delayed-rectifier potassium current IKs), LQT2 (KCNH2, loss of the rapid delayed-rectifier potassium current IKr), and LQT3 (SCN5A, gain of late sodium current INa). These three account for about seventy to seventy-five per cent of all genotype-positive LQTS. The rarer subtypes — LQT4 through LQT17 — involve ankyrin-B, the potassium channel accessory proteins (KCNE1, KCNE2), calcium-handling proteins (calsequestrin, calmodulin), and other structural proteins, and several present in infancy with severe phenotypes. [3] [11]
LQT1 (KCNQ1)
IKs loss
- Triggers: exercise, swimming, emotion
- Most responsive to beta-blockers
- Broad-based T waves on ECG
- Most common subtype (~40–45%)
- Swimming is relatively specific for LQT1
LQT2 (KCNH2)
IKr loss
- Triggers: sudden loud noises, postpartum
- Auditory stimuli (alarm clocks, phones)
- Low-amplitude notched T waves
- Moderate beta-blocker response
- Potassium levels matter — hypokalaemia worsens
LQT3 (SCN5A)
INa gain
- Triggers: sleep, rest, bradycardia
- Least responsive to beta-blockers
- Long ST segment with late-onset T wave
- May respond to mexiletine or ranolazine
- Higher event rate at lower heart rates
The related channelopathies round out the clinical picture. Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a calcium-handling disorder caused by RYR2 mutations in most cases, producing calcium leak from the sarcoplasmic reticulum during adrenergic stress; its resting ECG is normal and its hallmark is bidirectional or polymorphic VT on exercise. Brugada syndrome is a sodium-channel disorder (most commonly SCN5A loss of function) characterised by a coved ST-segment elevation in the right precordial leads and a risk of polymorphic VT and sudden death, typically presenting in adulthood. Short-QT syndrome is a rare gain-of-function potassium-channel disorder producing a QTc below 340 milliseconds with a risk of atrial and ventricular fibrillation. [4] [13] [14]
Epidemiology & Risk Factors
Congenital LQTS affects approximately one in two thousand individuals, though the true prevalence may be higher because mild or concealed cases are never detected. There is a female predominance in adulthood (though males present earlier), and the phenotype is highly variable even within families carrying the same mutation — penetrance is incomplete, and some genotype-positive individuals have a normal QTc (concealed LQTS). [1] [10]
The most important risk stratification variables are the corrected QT interval, the genotype, and the symptom history. A QTc above 500 milliseconds confers high risk of arrhythmic events regardless of genotype. Among the three major genotypes, LQT1 confers the lowest risk (and the best response to beta-blockers), LQT2 an intermediate risk, and LQT3 the highest risk per event (and the poorest beta-blocker response). A prior syncope or aborted cardiac arrest is the strongest clinical predictor of future events, and events are most frequent in the first decade of life for males and around puberty for both sexes. [5] [9]
The numbers that anchor your viva
Sudden cardiac death in the young is the presenting scenario that brings the channelopathies to the clinician's attention. The prospective Australian study of sudden cardiac death in children and young adults found that a substantial proportion had a clinically identifiable cardiac cause, and that inherited arrhythmia syndromes — including LQTS and CPVT — were among the most common findings when the heart was structurally normal. This underpins the recommendation that every unexplained syncope, seizure, or sudden death in a child or young adult warrants an ECG and, where indicated, a specialist inherited cardiac conditions assessment. [6]
Pathophysiology
The teaching model rests on a single concept: the action potential of the ventricular myocyte is governed by the co-ordinated flow of ions through specific channels, and a mutation that disrupts any one channel changes the duration and shape of the action potential, which in turn changes the QT interval on the surface ECG and the vulnerability to arrhythmia. [2] [12]
In LQT1, the loss of the slow delayed-rectifier potassium current (IKs) removes a key repolarising current, so the action potential prolongs. The IKs current is particularly important during sympathetic stimulation, because it normally shortens the action potential when the heart rate rises — this is why LQT1 patients are vulnerable during exercise, emotion, and particularly swimming. The channel cannot adapt to the adrenergic surge, repolarisation lags, and early afterdepolarisations emerge. In LQT2, the loss of the rapid delayed-rectifier potassium current (IKr) similarly prolongs repolarisation, but the trigger is different: sudden arousal and auditory stimuli, probably because the abrupt surge in catecholamines stresses the already-compromised IKr channel. In LQT3, a gain of late sodium current (INa) causes a prolonged plateau phase of the action potential, which is why events occur at slow heart rates during sleep or rest. [1] [10]
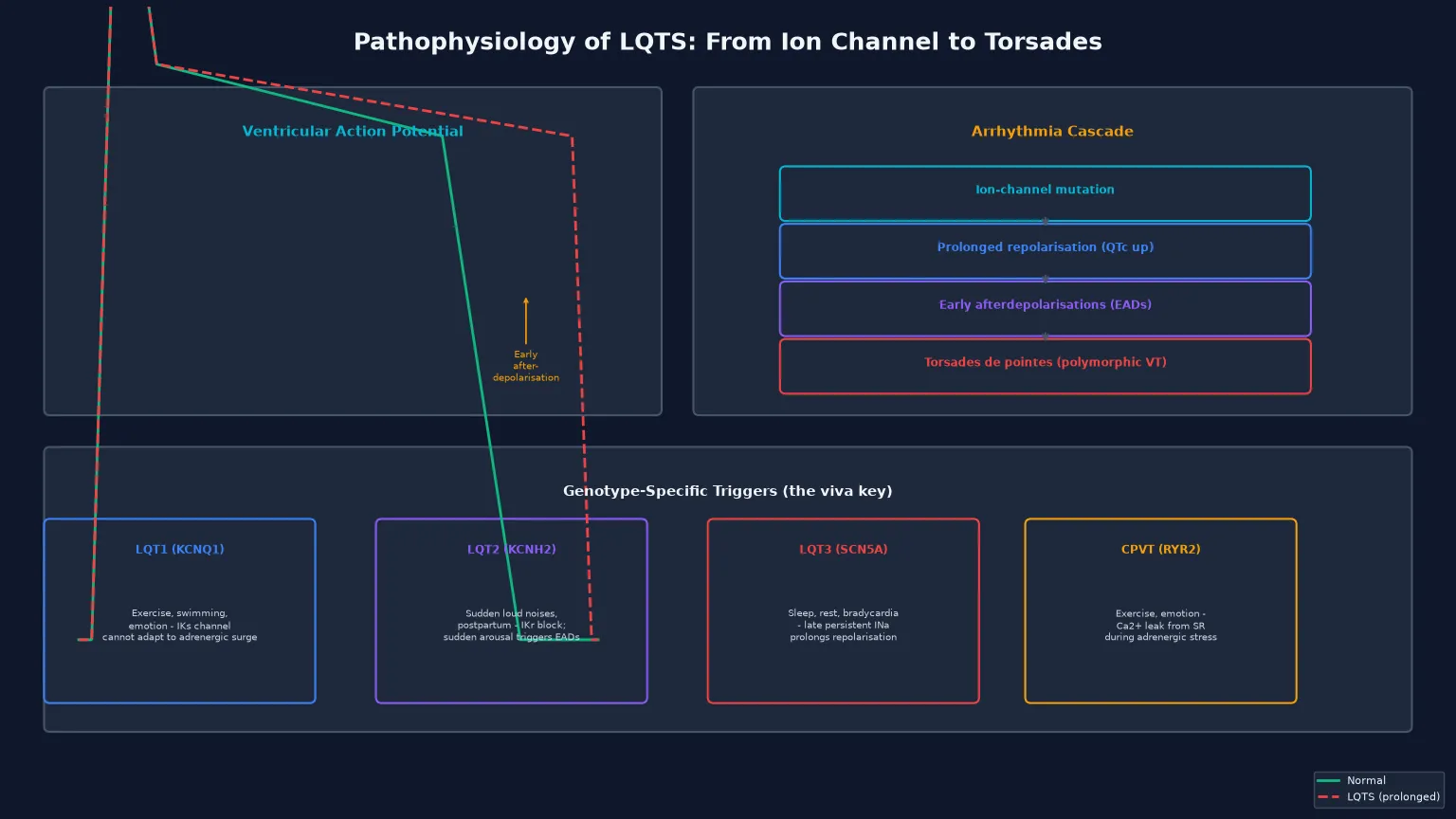
The early afterdepolarisation is the cellular event that bridges the prolonged QT interval to clinical arrhythmia. During the prolonged repolarisation, secondary depolarising oscillations appear during phases 2 and 3 of the action potential. When these reach threshold, they trigger ectopic beats that can initiate re-entry and the polymorphic ventricular tachycardia known as torsades de pointes. The torsades may terminate spontaneously (experienced as a syncope) or degenerate into ventricular fibrillation and sudden cardiac death. [2] [12]
[1] [4]In CPVT, the mechanism is entirely different and the resting ECG is normal. Mutations in the cardiac ryanodine receptor (RYR2) cause excessive calcium leak from the sarcoplasmic reticulum during diastole, particularly under adrenergic stimulation. The calcium overload produces delayed afterdepolarisations that trigger bidirectional or polymorphic VT during exercise or emotion. The heart is structurally normal, the resting ECG is normal, and only exercise testing — or a genetic test in a known family — reveals the disorder. [13]
Clinical Presentation
The presentation of LQTS ranges from an incidental finding on an ECG obtained for another reason, through unexplained syncope or seizure-like episodes, to sudden cardiac death. The age and context of presentation depend heavily on the genotype. [2] [6]
The classic presentations are syncope during or immediately after exertion (LQT1), syncope triggered by sudden loud noises such as an alarm clock or a ringing telephone (LQT2), and syncope or nocturnal events during sleep or rest (LQT3). Seizure-like movements are common during the arrhythmic syncope, which is why LQTS is a well-recognised cause of misdiagnosed epilepsy — a child may be treated with anticonvulsants for years before the QT interval is measured. A family history of syncope, sudden death, or "seizures" in first-degree relatives is a critical clue, as is a history of deafness (Jervell and Lange-Nielsen syndrome, the autosomal recessive cardio-auditory form of LQTS). [1] [10]
Neonatal and infantile presentations deserve particular attention. Severe neonatal LQTS may present with bradycardia due to 2:1 atrioventricular block (the QT is so prolonged that every other P wave conducts), with foetal bradycardia, or with sudden infant death. Calmodulin mutations (LQT14–16) are a recently recognised cause of recurrent cardiac arrest in infants, with extremely prolonged QTc and poor response to conventional therapy. [11] [9]
| Clinical picture | What it implies | Act |
|---|
Differential Diagnosis
Build the differential in two layers: first, the causes of a prolonged QT interval other than congenital LQTS, and second, the causes of syncope or sudden death in a child that mimic a channelopathy. The distinction is critical because the management differs fundamentally. [2] [4]
The acquired causes of QT prolongation are numerous and must be excluded before committing to a diagnosis of congenital LQTS. Electrolyte disturbances — particularly hypokalaemia, hypocalcaemia, and hypomagnesaemia — prolong the QT interval and are common in sick children. A long list of medications prolong the QT interval, including macrolide and fluoroquinolone antibiotics, antifungals, antiemetics (ondansetron), antipsychotics, methadone, and certain antihistamines. Eating disorders, liquid protein diets, and intracranial events (stroke, intracranial haemorrhage) are further causes. Every child with a newly prolonged QTc needs a medication review and an electrolyte panel before a genetic diagnosis is pursued. [4] [15]
Acquired QT prolongation
- Electrolyte disturbance (low K+, Ca2+, Mg2+)
- QT-prolonging drugs (macrolides, ondansetron, antipsychotics, methadone)
- Eating disorders, liquid protein diets
- Intracranial events
- Resolves when the cause is corrected
Syncope mimics
- Vasovagal syncope (prodrome, typical triggers, rapid recovery)
- Breath-holding attacks in toddlers
- Orthostatic hypotension (adolescent growth spurts)
- Post-concussive syncope
- True epilepsy (prodrome, post-ictal state)
SCD mimics in structurally normal heart
- CPVT (normal resting ECG, exercise-provoked VT)
- Brugada syndrome (coved ST in V1-V3)
- Short-QT syndrome (QTc < 340 ms)
- Wolff-Parkinson-White (pre-excitation, accessory pathway)
- Myocarditis (may be transiently structurally normal)
The key to resolving the syncope differential is the history and the twelve-lead ECG. Vasovagal syncope has a characteristic prodrome (warmth, nausea, visual disturbance), occurs in standing or hot environments, and recovers rapidly. Arrhythmic syncope is typically sudden, without prodrome, and may occur during exertion or in water. A careful family history of syncope, sudden death, or deafness is non-negotiable. The ECG should be obtained in every child presenting with syncope, and the QT interval should be measured manually — not relied upon from the automated report, which is frequently inaccurate in children. [2] [1]
Clinical & Bedside Assessment
Assessment runs from the history through to a careful twelve-lead ECG with manual QT measurement, because the diagnosis hinges on the electrocardiogram and the clinical story. A child presenting after a syncopal episode needs a focused cardiac assessment before being labelled with a neurological or vasovagal diagnosis. [1] [6]
The history targets the circumstances of the event: was it exertional, emotional, nocturnal, or triggered by a loud noise? Was there a prodrome (suggesting vasovagal) or was it sudden without warning (suggesting arrhythmia)? How rapid was the recovery? Is there a family history of syncope, sudden death, seizures, deafness, or known LQTS? Has the child been taking any QT-prolonging medications? The collateral history from witnesses is invaluable, because the child may not recall the event. [2] [10]
[1] [4]The ECG assessment requires manual measurement of the QT interval from the onset of the QRS complex to the end of the T wave, using the lead with the clearest T-wave termination (usually lead II or V5), and correction for heart rate using Bazett's formula (QTc = QT / square root of the RR interval in seconds). Bazett's formula over-corrects at high heart rates and under-corrects at low heart rates, so in children (who have faster heart rates) the Fridericia or Framingham corrections may be more accurate. A QTc above 460 milliseconds in children (above 470 in post-pubertal males, above 480 in post-pubertal females) is considered prolonged, and a QTc above 500 milliseconds is diagnostic of LQTS regardless of symptoms. [1] [15]
Investigations
The diagnostic strategy has three tiers: the resting twelve-lead ECG with the Schwartz diagnostic score, provocative testing (exercise or epinephrine challenge) for concealed cases, and genetic testing for confirmation and cascade screening. Echocardiography is performed to exclude structural heart disease, because LQTS is a diagnosis of the structurally normal heart. [1] [3]
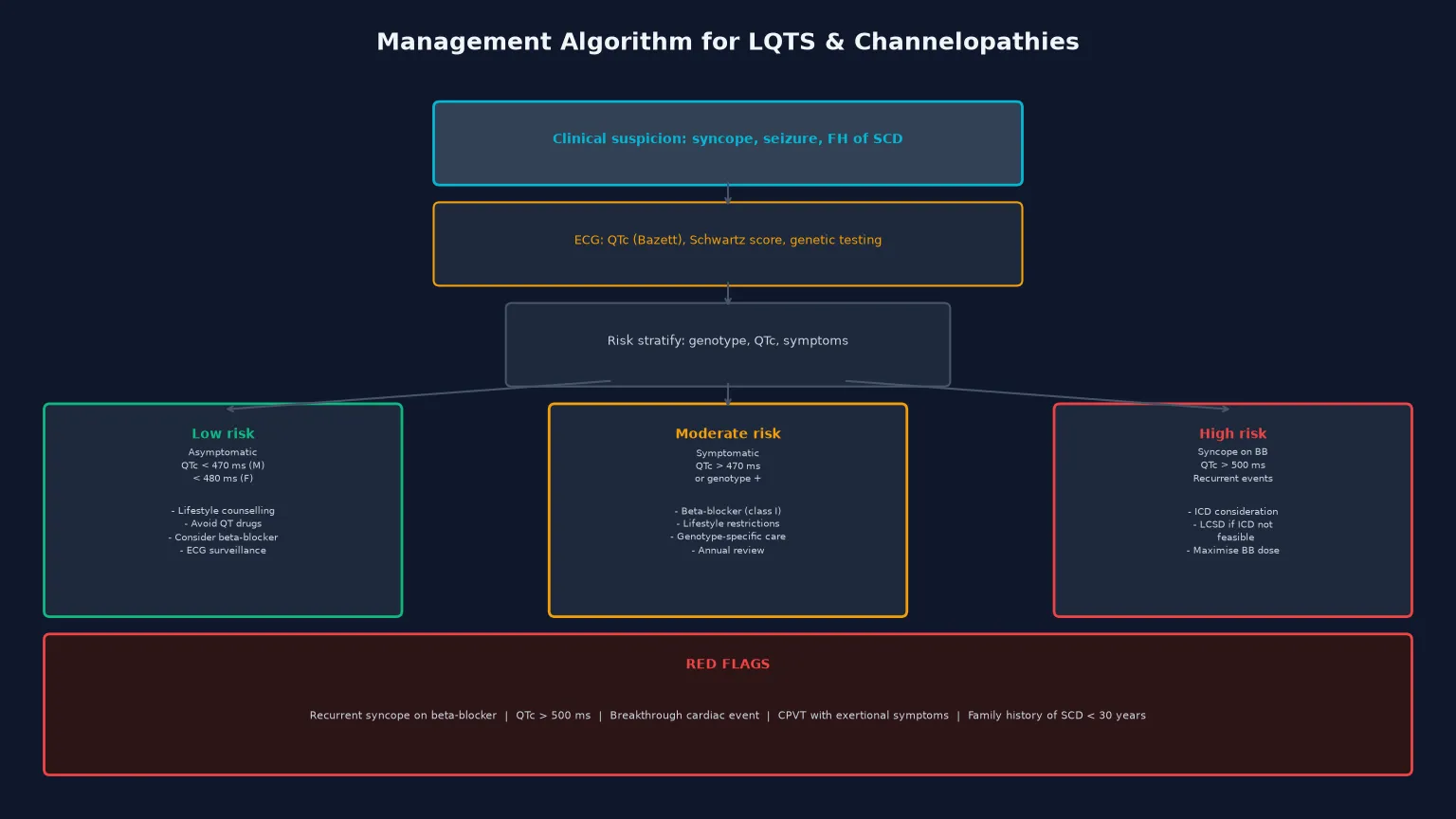
The Schwartz diagnostic score integrates the QTc, the clinical history, and the family history into a probabilistic assessment that guides the intensity of investigation. Points are awarded for QTc intervals (with higher scores for longer intervals), for torsades de pointes, for syncope with specific triggers (exercise, emotional stress), for congenital deafness, and for a family history of definite LQTS or unexplained sudden death before age thirty in an immediate family member. A score of three or fewer points is low probability, three-and-a-half to intermediate, and four or more is high probability, warranting genetic testing. [1] [4]
Exercise testing and epinephrine challenge are useful for concealed LQTS, in which the resting QTc is normal but the channel cannot adapt to sympathetic stress. During exercise testing, the QTc fails to shorten appropriately with exertion and recovery in LQT1, and paradoxical QT prolongation during low-dose epinephrine infusion is characteristic of LQT1 (and distinguishes it from LQT2 and LQT3). In CPVT, exercise testing is the key diagnostic test, because the resting ECG is normal and bidirectional or polymorphic VT appears only during adrenergic stress. [7]
The standard diagnostic workup
12-lead ECG with manual QTc measurement (Bazett, confirmed with Fridericia in children); assess T-wave morphology.
Schwartz diagnostic score integrating QTc, clinical history, and family history.
Echocardiography to exclude structural heart disease (LQTS is a structurally normal heart diagnosis).
Exercise stress test if LQT1 or CPVT suspected — assess QT adaptation and look for bidirectional VT.
Epinephrine QT stress testing for concealed LQTS (paradoxical QT prolongation in LQT1).
Genetic testing (KCNQ1, KCNH2, SCN5A first; expanded panel for rarer subtypes) through an inherited cardiac conditions service.
Cascade screening of all first-degree relatives with ECG and, if a mutation is found, targeted genetic testing.
Genetic testing confirms the diagnosis, identifies the genotype (which guides therapy), and enables cascade screening of relatives. The yield of genetic testing is about seventy to seventy-five per cent for the three major genes (KCNQ1, KCNH2, SCN5A), and expanded panels include the rarer subtypes. A pathogenic mutation confirms the diagnosis and allows predictive testing of relatives; a negative genetic test does not exclude LQTS, because some mutations are not detected by current sequencing. Genetic counselling is essential before and after testing, because the result has implications for the individual and for every first-degree relative. [3] [12]
Management — Resuscitation
The acute arrhythmic event in LQTS is torsades de pointes, which may present as syncope, seizure-like activity, cardiac arrest, or sudden death. The resuscitation follows the standard paediatric life support algorithm with several channelopathy-specific modifications. [4] [9]
For a child presenting with torsades de pointes, defibrillate if the rhythm is pulseless (the paediatric advanced life support protocol applies). If the torsades is sustained but the child has a pulse, give intravenous magnesium sulphate (25–50 mg/kg, up to 2 g), which suppresses early afterdepolarisations and is the most effective acute therapy for torsades. Correct any electrolyte abnormalities (particularly potassium and magnesium), and discontinue all QT-prolonging medications. Temporary overdrive pacing (atrial or ventricular) at a rate of 100–120 beats per minute shortens the QT interval and suppresses torsades by reducing the action-potential duration. Isoprenaline may be used to maintain a faster heart rate while awaiting definitive management. [9] [4]
TORSADES-BREAK
For the child with known LQTS presenting with a breakthrough event on therapy, the message is escalation, not dose-doubling: assess adherence, confirm the genotype (LQT3 responds poorly to beta-blockers), and escalate to left cardiac sympathetic denervation or an implantable cardioverter defibrillator. The child with CPVT in ventricular fibrillation is resuscitated with the standard protocol, and intravenous beta-blockade is given early, because the arrhythmia is catecholamine-driven. [8] [13]
Management — Definitive & Stepwise
Definitive management is stratified by risk and guided by genotype, and the modern approach has four pillars: lifestyle modification, beta-blockade, non-pharmacological adjuncts (left cardiac sympathetic denervation), and the implantable cardioverter defibrillator for the highest-risk patients. The goal is to reduce arrhythmic events while minimising the burden and complications of therapy, particularly in children. [5] [9]
Lifestyle modification is universal for all genotype-positive patients regardless of symptoms or QTc. Competitive and high-intensity sports are restricted (the 2013 consensus and the 2015 ESC guidelines both moved toward permitting some low-intensity sport for well-treated patients, but competitive and exertional sports remain restricted for most). QT-prolonging medications are avoided for life — every patient and family should be given a current list and counselled to check before taking any new prescription. Hypokalaemia and dehydration are corrected aggressively during intercurrent illness. [4]
Beta-blockers are the first-line pharmacological therapy for all symptomatic patients and for most genotype-positive asymptomatic patients. Propranolol (2–4 mg/kg/day in divided doses) and nadolol (0.5–2 mg/kg/day, once or twice daily) are the most commonly used agents in children; nadolol has emerged as the preferred agent because of its long half-life, tolerability, and possibly superior efficacy. The landmark study by Moss and colleagues demonstrated that beta-blockers reduce arrhythmic events by approximately two-thirds across the LQTS cohort, but the protection is genotype-dependent: LQT1 patients are well-protected, LQT2 patients have intermediate protection, and LQT3 patients derive the least benefit — this genotype-dependent efficacy is one of the most important teaching points. [5] [9]
The life arc of a child with long-QT syndrome
For the high-risk subset — those with breakthrough events on maximally tolerated beta-blockers, a QTc above 500 milliseconds, or a history of aborted cardiac arrest — left cardiac sympathetic denervation (LCSD) and the implantable cardioverter defibrillator (ICD) are the escalation options. LCSD is a thoracoscopic procedure that removes the lower half of the left stellate ganglion and the first three to four thoracic ganglia, reducing cardiac sympathetic input without the complications of an implanted device. Schwartz and colleagues demonstrated that LCSD produces a greater than ninety per cent reduction in arrhythmic events and is particularly valuable in children in whom ICD implantation is technically challenging or psychologically burdensome. LCSD is now recommended as a first-line non-pharmacological adjunct for patients with recurrent events on beta-blockers who do not yet meet ICD criteria, and as an adjunct to the ICD to reduce shocks. [8] [9]
The ICD is reserved for the highest-risk patients: survivors of aborted cardiac arrest, those with recurrent syncope despite beta-blockers and LCSD, and those with very high-risk genotypes or phenotypes. The threshold for ICD implantation in children is higher than in adults because of the technical challenges (lead fractures, growth-related complications, infection) and the psychological burden of inappropriate shocks, which are particularly common in young patients with LQTS who may have rapid physiological heart rates that overlap with the ventricular fibrillation detection zone. The decision is individualised and made in a specialist inherited cardiac conditions multi-disciplinary team. [9] [4]
Specific Subtypes & Scenarios
LQT1 (KCNQ1) is the most common and most treatable subtype, with events typically triggered by exercise, swimming, and emotional stress. Beta-blockers are highly effective, and the prognosis on therapy is excellent. The ECG shows broad-based T waves. The diagnosis is confirmed by genetic testing; in concealed cases (normal resting QTc), epinephrine challenge shows paradoxical QT prolongation. Swimming-related symptoms are relatively specific for LQT1 and mandate investigation. [1] [7]
LQT2 (KCNH2) is triggered by sudden loud noises and the postpartum period, and its ECG signature is low-amplitude, notched (bifid) T waves. Hypokalaemia worsens the phenotype, so maintaining serum potassium above 4.0 mmol/L is part of management. Beta-blockers are moderately effective, and potassium supplementation may be beneficial. Postpartum women with LQT2 are at particularly high risk in the first nine months after delivery and require intensified surveillance. [10] [4]
LQT3 (SCN5A) presents with events during sleep or rest and has a long, flat ST segment with a late-onset T wave on the ECG. Beta-blockers are less effective (because the mechanism is a gain of sodium current rather than adrenergic sensitivity), and mexiletine or ranolazine — late sodium-current blockers — may be used as adjuncts to shorten the QT interval. The risk per event is higher than in LQT1, so the ICD threshold is lower. [2] [9]
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a distinct entity with a structurally normal heart, a normal resting ECG, and exercise- or emotion-triggered bidirectional or polymorphic VT. It is caused by mutations in the cardiac ryanodine receptor (RYR2, autosomal dominant, approximately sixty to seventy per cent of cases) or calsequestrin (CASQ2, autosomal recessive), and more recently described calmodulin and triadin mutations cause severe infantile forms. Management is beta-blockade (nadolol is first-line), with flecainide as an adjunct for breakthrough ventricular ectopy, and LCSD or ICD for recurrent events. CPVT has a high event rate if untreated and a well-described risk of sudden death in childhood, so exercise testing is mandatory for any child with exertional syncope and a normal resting ECG. [13] [12]
Brugada syndrome is rare in children and typically presents in adulthood, but paediatric cases occur, particularly in families with known SCN5A mutations. The characteristic ECG shows coved ST-segment elevation in the right precordial leads (V1–V3), which may be concealed and revealed by sodium-channel blockade (ajmaline or flecainide challenge) or by fever. The risk is polymorphic VT and sudden death, and management is ICD implantation for symptomatic patients, with fever treated aggressively because it can unmask the ECG and trigger arrhythmia. The Shanghai Score System integrates ECG, clinical, and genetic features to guide diagnosis and risk stratification. [14]
Short-QT syndrome is the rarest of the channelopathies, defined by a QTc below 340 milliseconds (though diagnostic thresholds vary by QTc formula). It is caused by gain-of-function potassium-channel mutations (KCNH2, KCNQ1, KCNJ2) that abbreviate repolarisation, and it presents with atrial fibrillation, syncope, or sudden death. Management is quinidine (which prolongs the QT interval) or an ICD. The condition is easily missed because a short QT is often overlooked, and the choice of QTc formula affects the measured interval — this is a diagnostic pitfall worth knowing. [15]
Complications & Pitfalls
The complications of LQTS are the arrhythmic events themselves — syncope, aborted cardiac arrest, and sudden cardiac death — and the complications of therapy. On the disease side, untreated LQTS has an estimated annual mortality of about one per cent per year, with the highest risk in those with prior symptoms, a QTc above 500 milliseconds, and the LQT2 or LQT3 genotype. [5] [9]
On the therapy side, beta-blockers are generally well tolerated in children but may cause fatigue, exercise intolerance, bronchospasm, hypoglycaemia (particularly in infants), and mood changes. Poor adherence is the commonest reason for breakthrough events, and a frank discussion about adherence is part of every follow-up visit. The ICD brings a distinct set of complications that are more burdensome in children than in adults: lead fractures (from growth and activity), infection, inappropriate shocks (the paediatric heart rate overlaps with detection zones), and the psychological impact of living with a device. Left cardiac sympathetic denervation has a lower complication profile (Horner's syndrome in a small percentage, transient) and is increasingly favoured as an intermediate step. [8] [9]
The risks that drive management intensity
The common clinical pitfalls are: failing to measure the QT interval in a child with syncope or seizure (the single most common error); relying on the automated QTc from the ECG machine without manual confirmation; misdiagnosing arrhythmic syncope as epilepsy and treating with anticonvulsants; failing to exclude acquired causes of QT prolongation (electrolytes, medications) before pursuing a genetic diagnosis; under-dosing or poor adherence with beta-blockers; and failing to cascade-screen first-degree relatives after a diagnosis. The psychosocial burden of an inherited arrhythmia diagnosis — anxiety, restrictions, the impact on siblings and parents — is also a pitfall if unaddressed. [2] [6]
Prognosis & Disposition
With modern management, the prognosis of LQTS is excellent for most patients. Beta-blocker therapy reduces events by roughly two-thirds, and the addition of LCSD and ICD therapy for the high-risk subset further reduces mortality. The estimated annual mortality on appropriate therapy is well below one per cent, and most patients lead full and active lives with appropriate restrictions and surveillance. [5] [10]
The prognosis is not uniform, however, and certain factors confer higher risk: a QTc above 500 milliseconds, the LQT2 or LQT3 genotype, a history of syncope or aborted cardiac arrest before therapy, and events within the first year of life. These patients need more intensive therapy (maximal beta-blockade, LCSD, and often an ICD) and closer surveillance. CPVT carries a high untreated event rate and requires aggressive management with beta-blockers and adjuncts. [9] [13]
Disposition is outpatient management under a specialist inherited cardiac conditions service, with the general paediatrician playing a critical role in co-ordination, adherence support, and the management of intercurrent illness (which can destabilise the QT interval through fever, electrolyte disturbance, and QT-prolonging medications). The adolescent needs structured transition to an adult inherited cardiac conditions service, with counselling about pregnancy (which is generally safe on beta-blockers but carries a transiently increased risk in the postpartum period, particularly in LQT2), contraception, and the heritable nature of the condition. [4] [10]
Special Populations
Neonates and infants with LQTS present particular challenges. Severe congenital LQTS may present with foetal bradycardia, 2:1 atrioventricular block (the QT is so prolonged that alternate P waves fall into the refractory period), or sudden infant death. Calmodulin mutations cause a particularly severe infantile phenotype with extremely prolonged QTc, recurrent cardiac arrest, and poor response to conventional therapy. Beta-blocker dosing in infants requires attention to hypoglycaemia and feeding, and nadolol is preferred for its tolerability. [11] [9]
Children with epilepsy or developmental delay who are on multiple medications are at risk of both congenital LQTS being misdiagnosed as epilepsy and of acquired QT prolongation from anticonvulsants, antipsychotics, and other agents. Every child on a QT-prolonging medication deserves periodic ECG surveillance, and every child with "atypical" or treatment-resistant epilepsy deserves a baseline ECG. [2] [6]
The transitioning adolescent and young adult faces the convergence of several risks: adherence challenges, the highest-risk period for events (puberty and the early twenties), recreational drug and alcohol use, and the transition to adult care. Structured transition programmes — education, self-management, and a warm handover to the adult inherited cardiac conditions service — are now standard and are emphasised in the HRS/EHRA/APHRS and CSANZ guidance. Pregnancy planning is a key element, because pregnancy is generally safe on beta-blockers but the postpartum period is a high-risk window, particularly in LQT2. [4] [10]
Resource-limited and remote populations — including Indigenous, refugee, and rural communities — face diagnostic delays and limited access to genetic testing and specialist services. Telehealth cardiology, protocol-driven beta-blocker management, and a low threshold for referral after any exertional syncope are part of equitable practice. The global reality is that most children with LQTS are never diagnosed, and the index presentation in many families is a sudden death that could have been prevented by a twelve-lead ECG. [6] [9]
Evidence, Guidelines & Regional Differences
| Region | Key guideline | First-line therapy | ICD / LCSD threshold |
|---|
The controversies a candidate should be able to discuss are the intensity of sports restriction (the 2013 consensus moved toward permitting some sport for well-treated patients, a shift from the traditional blanket ban), the role of LCSD versus the ICD in children (LCSD is increasingly favoured as the intermediate step to avoid device complications), the management of genotype-negative LQTS (clinical diagnosis with the Schwartz score, treated as genotype-positive), and the approach to the asymptomatic genotype-positive child with a borderline QTc (individualised, but most guidelines recommend beta-blockers for KCNQ1 and KCNH2 carriers). [4] [9]
Exam Pearls
References
- [1]Schwartz PJ, Moss AJ, Vincent GM, Crampton RS Diagnostic criteria for the long QT syndrome. An update. Circulation, 1993.PMID 8339437
- [2]Abrams DJ, Macrae CA Long QT syndrome. Circulation, 2014.PMID 24709866
- [3]Napolitano C, Priori SG, Schwartz PJ, Block RW, Taroni F, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA, 2005.PMID 16414944
- [4]Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm, 2013.PMID 24011539
- [5]Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation, 2000.PMID 10673253
- [6]Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med, 2016.PMID 27332903
- [7]Vyas H, Hejlik J, Ackerman MJ Epinephrine QT stress testing in congenital long QT syndrome. Circulation, 2006.PMID 16962127
- [8]Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation, 2004.PMID 15051644
- [9]Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace, 2015.PMID 26318695
- [10]Goldenberg I, Moss AJ Long QT syndrome. J Am Coll Cardiol, 2008.PMID 18549912
- [11]Crotti L, Johnson CN, Graf E, De Marchi GM, Rohr CF, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation, 2013.PMID 23388215
- [12]Cerrone M, Priori SG A clinical approach to inherited arrhythmias. Circ Cardiovasc Genet, 2012.PMID 23074337
- [13]Bhuiyan ZA, van den Werf C, Henkens M, Jordaens L, Vatta M, et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation, 2007.PMID 17875969
- [14]Kawada S, Morita H, Antzelevitch C, Morita ST, Kaneko M, et al. Shanghai Score System for Diagnosis of Brugada Syndrome: validation of the score system and system reclassification of patients. JACC Clin Electrophysiol, 2018.PMID 29929664
- [15]Providência R, Faustino A, Ferreira MJ, Gonçalves L, Trigo J, et al. Impact of QTc formulae in the prevalence of short corrected QT interval and impact on probability of diagnosis of short QT syndrome. Heart, 2018.PMID 28954836