Paeds · cardiology
Pulmonary hypertension in children
Also known as pulmonary hypertension · pulmonary arterial hypertension · PAH · paediatric pulmonary hypertension · Eisenmenger syndrome · idiopathic pulmonary arterial hypertension · IPAH
A fellowship approach to pulmonary hypertension in children: a mean pulmonary artery pressure above twenty is never normal, the WHO group decides the cause, the cardiac catheter measures the resistance, and the right ventricle decides the prognosis. The thread runs from the syncope or the loud second heart sound through the echocardiogram and the catheter to combination therapy with phosphodiesterase-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues, and on to the surgical escalations of septostomy, the Potts shunt, and transplantation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A four-year-old is brought in after collapsing while running, with an examination that reveals a loud pulmonary component of the second heart sound; or a teenager with a ventricular septal defect repaired late now has cyanosis and a decreasing exercise tolerance, the Eisenmenger physiology; or an ex-preterm infant with bronchopulmonary dysplasia fails to wean from oxygen and has a rising tricuspid regurgitation velocity on echocardiography. The fellowship task in each is the same: recognise the raised pulmonary pressure, confirm it at catheter, assign the group, and protect the right ventricle. [5] [9]
The five moves — Suspect, Scan, Catheterise, Classify, Escalate
Overview & Definition
Pulmonary hypertension is defined haemodynamically at cardiac catheterisation as a mean pulmonary artery pressure above twenty millimetres of mercury. The sixth World Symposium refined this in 2018, and the 2022 European guideline carries the same threshold, separating pulmonary arterial hypertension from the other groups by adding a raised resistance and a normal wedge pressure. [3] [12]
Pulmonary arterial hypertension, the first group, requires a pulmonary artery wedge pressure of fifteen or less and a pulmonary vascular resistance above two Wood units in adults, with an indexed resistance above three Wood units per square metre in children. The wedge pressure separates a problem in the pulmonary arteries, where it is normal, from a problem downstream in the left heart, where it is raised. This single number is the safeguard against giving a vasodilator to a child whose true problem is left heart disease. [2] [1]
The condition is distinguished from persistent pulmonary hypertension of the newborn, which is a transitional circulation problem of the first days of life and is addressed on its own page. The child with established pulmonary hypertension presents weeks, months, or years later, and the resistance is fixed rather than transitional. The distinction matters because the management, the prognosis, and the counselling diverge sharply between the two. [5] [1]
Classification
Classification follows the World Health Organization clinical system, which sorts pulmonary hypertension into five groups by the underlying cause, because the group decides the investigation pathway and the therapy. The Panama modification adapts the system for children, emphasising congenital heart disease, developmental lung disease, and syndromic causes that dominate the paediatric caseload. [3] [2]
Group one is pulmonary arterial hypertension. It holds the idiopathic and heritable forms, drug and toxin disease, and the associated causes, of which congenital heart disease is the commonest in children. Heritable disease runs through bone morphogenetic protein receptor type 2 and other mutations, and the idiopathic and heritable forms are the ones tested for vasoreactivity at catheterisation. Persistent pulmonary hypertension of the newborn sits in a primed group, a transitional disease distinct from the fixed resistance of the older child. [2] [5]
Group two is pulmonary hypertension from left heart disease, where a raised left atrial pressure is transmitted backwards. Group three is lung disease or hypoxia, and in children this is dominated by bronchopulmonary dysplasia, developmental lung hypoplasia such as congenital diaphragmatic hernia, cystic fibrosis, and sleep-disordered breathing. Group four is pulmonary artery obstruction, chiefly chronic thromboembolic disease, and group five holds the multifactorial and unclear causes, where complex congenital lesions and systemic diseases sit. [3] [9]

Epidemiology & Risk Factors
The idiopathic and heritable form of pulmonary arterial hypertension affects roughly two children per million, and the congenital-heart-disease-associated form is several times more common. The Tracking Outcomes and Practice in Paediatric Pulmonary Hypertension registry, the largest prospective paediatric cohort, showed that congenital heart disease accounts for the majority of paediatric cases, followed by idiopathic and heritable disease. [6] [7]
The risk factors that matter for the fellowship answer are the conditions that scar the pulmonary bed early. Bronchopulmonary dysplasia is the dominant cause in the ex-preterm infant, with up to a quarter of severely affected survivors showing echocardiographic pulmonary hypertension, and the disease carries a measurable mortality. Congenital diaphragmatic hernia and other causes of developmental lung hypoplasia produce pulmonary hypertension through a small vascular bed, and the scimitar syndrome and the unrepaired left-to-right shunt do so through over-circulation. [9] [5]
Down syndrome carries a striking burden of pulmonary hypertension through several routes — persistent pulmonary hypertension of the newborn, developmental lung hypoplasia, upper airway obstruction, and the unrepaired atrioventricular septal defect. A fellowship candidate should screen the cardiovascular status of every child with Down syndrome and not assume the murmur is benign. The heritable form runs through bone morphogenetic protein receptor type 2 and through the genes of the smaller associated pathways, and genetic testing and counselling are now part of the work-up of idiopathic disease. [1] [2]
The natural history has improved markedly with modern therapy, but the untreated idiopathic form remains a life-limiting disease. The risk-stratified approach, which sorts children into low, intermediate, and high risk by clinical, echocardiographic, and biochemical markers, replaced the older binary framing and now drives the intensity of treatment. A high-risk child is escalated to parenteral prostacyclin and evaluated for transplantation. [4] [5]
Pathophysiology
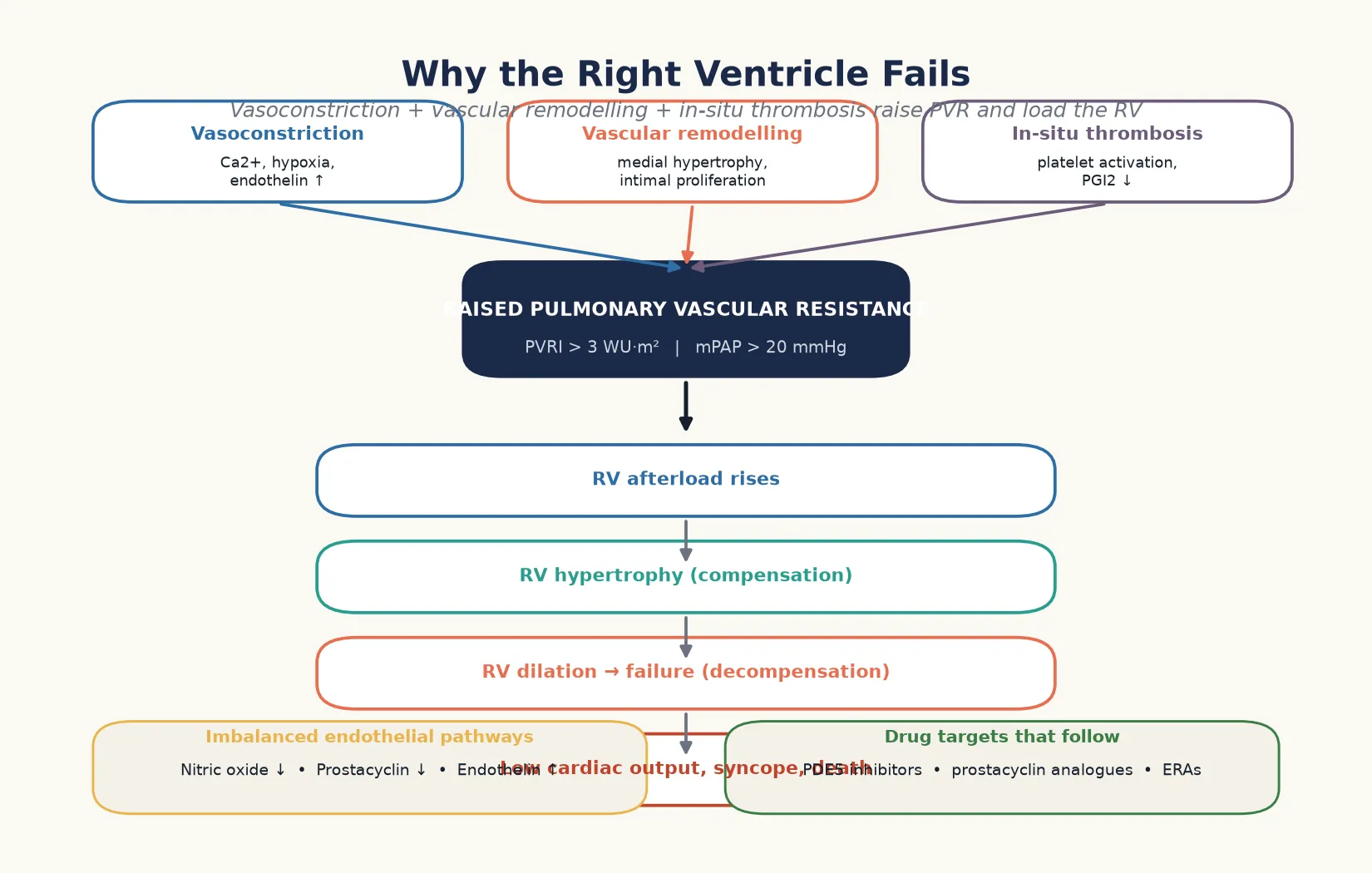
The pathophysiology turns on a pulmonary vascular bed that has narrowed, so the right ventricle pumps against a higher afterload. Three forces drive the narrowing, and they are the targets of the three drug pathways. [1] [5]
Vasoconstriction narrows the vessels through smooth muscle contraction, driven by hypoxia, calcium, and the excess of vasoconstrictors over vasodilators. Vascular remodelling thickens the vessel wall through medial hypertrophy and intimal proliferation, so the lumen narrows even when the muscle relaxes. In-situ thrombosis narrows it further through platelet aggregation and microthrombi. The three together raise the resistance, and the resistance, not the pressure alone, is what the right ventricle feels. [1] [12]
The endothelium sits at the centre of the imbalance. Nitric oxide and prostacyclin, the two vasodilators, fall, while endothelin, the vasoconstrictor and mitogen, rises. The drugs restore this balance from three directions — phosphodiesterase-5 inhibitors protect the nitric oxide that remains, prostacyclin analogues replace what is lost, and endothelin receptor antagonists block the excess. This is the logic of combination therapy, which treats the disease from all three angles rather than relying on a single agent. [4] [2]
The right ventricle adapts first and fails later. It hypertrophies to generate the higher pressure, and the hypertrophy is the compensation. When the afterload exceeds its capacity, the ventricle dilates, the tricuspid regurgitation worsens, the cardiac output falls, and the child presents with syncope, exertional intolerance, and signs of right heart failure. This is why the right ventricle decides the prognosis, and why a child in right ventricular failure is escalated urgently. [5] [1]
Clinical Presentation
The presentation differs with age, and the fellowship skill is to hear the subtle early story rather than wait for the dramatic late one. An infant presents with poor feeding, sweating with feeds, tachypnoea, and failure to thrive, the picture of a pressure-loaded right ventricle struggling to deliver a cardiac output. An older child presents with exertional dyspnoea, fatigue, chest pain, and the red-flag symptom of syncope. [5] [2]
Syncope on exertion is the single most important symptom, because it signals a cardiac output that cannot rise with demand. A child who faints while running has pulmonary hypertension until the echocardiogram proves otherwise, and the same applies to presyncope and exertional chest pain. The onset is often gradual, and families attribute the fatigue to deconditioning or asthma before the loud second heart sound is heard. [1] [5]
Examination holds the bedside signature. The loud, sometimes palpable pulmonary component of the second heart sound is the hallmark of a raised pulmonary pressure, and it is the finding that should trigger the echocardiogram. A right ventricular heave, a pansystolic tricuspid regurgitation murmur at the lower left sternal edge, and an early diastolic pulmonary regurgitation murmur complete the picture. Jugular venous distension, hepatomegaly, and peripheral oedema signal right heart failure, and cyanosis appears when an intracardiac shunt allows right-to-left flow. [1] [11]
The Eisenmenger presentation is the late form of an unrepaired congenital shunt. A child with a ventricular septal defect or an atrioventricular septal defect that was repaired late, or never repaired, develops irreversible pulmonary vascular disease, and the shunt reverses from left-to-right to right-to-left. The cyanosis, the clubbing, and the exertional dyspnoea declare the physiology, and the once-operable lesion is no longer operable. Recognising the threshold — the resistance below which repair is safe and above which it is not — is the must-not-miss judgement. [8] [5]
Differential Diagnosis
The differential splits into two bedside questions. What else causes exertional syncope in a child, and what else causes a loud second heart sound with right heart strain? The discriminating move in both is the echocardiogram, which estimates the pulmonary pressure and assesses the right ventricle. [1] [11]
For the child with exertional syncope, the dangerous mimics are arrhythmia, hypertrophic cardiomyopathy, aortic stenosis, and the neurally mediated faint. The history discriminates: a neurally mediated faint is situational and brief, whereas syncope on peak exertion without warning fits a cardiac cause. The electrocardiogram and the echocardiogram close most of the differential, and a holter monitor captures the intermittent arrhythmia. [5] [1]
For the child with a loud second heart sound, the cause may be a thin chest wall that transmits the sound normally, but persistence after the first few months, or association with a right ventricular heave, earns the echocardiogram. The other causes of right heart strain — a large left-to-right shunt before the resistance rises, an atrial septal defect with right ventricular volume load, and pulmonary valve stenosis — declare their anatomy on the echocardiogram. The cardiac catheter separates the fixed resistance of established disease from the reversible flow load. [11] [2]
The group-level differential is the deeper question once the pressure is confirmed. Is the cause in the pulmonary arteries, the left heart, the lungs, the thromboembolic bed, or multifactorial? The work-up is directed by the group — an echocardiogram for the left heart, lung function and imaging for the lungs, a ventilation-perfusion scan for thromboembolic disease, and the sleep study for sleep-disordered breathing. The catheter, the wedge pressure, and the resistance close the group. [3] [12]
| Feature | Pulmonary hypertension | Atrial septal defect | Pulmonary stenosis | Innocent thin chest |
|---|---|---|---|---|
| P2 intensity | Loud, palpable | Wide, fixed split | Soft or absent | Normal, varies |
| Right ventricle | Pressure-loaded | Volume-loaded | Pressure-loaded | Normal |
| Murmur | TR / PR flow | Ejection systolic, pulmonary flow | Ejection systolic, ejection click | Vibratory, functional |
| Echo TR velocity | Raised | Normal or mildly raised | Normal | Normal |
| Key clue | Exertional syncope | Fixed split S2 | Ejection click, soft P2 | Otherwise well child |
Clinical & Bedside Assessment
The recognition move begins with the history, because the symptoms of early pulmonary hypertension are subtle and easy to dismiss. Ask about exercise tolerance, because fatigue and breathlessness on running are the earliest signs, and ask specifically about syncope and presyncope, because these are the red-flag symptoms. Ask about feeding and growth in the infant, because sweating with feeds and failure to thrive are the infant equivalents. Ask about a family history of pulmonary hypertension or sudden death, because the heritable form is relevant to counselling. [5] [1]
Examination is focused on the right heart and its consequences. Listen for the loud pulmonary component of the second heart sound, the single most reliable bedside finding, and feel for the right ventricular heave. Auscultate for the tricuspid and pulmonary regurgitation murmurs, and assess the jugular venous pressure, the liver size, and the perfusion, because these gauge the right ventricular function. Measure the oxygen saturation, because desaturation on exercise or at rest signals a right-to-left shunt, and inspect for clubbing, which declares long-standing cyanosis. [1] [11]
The functional capacity is graded, and the grade is part of the risk stratification. The World Health Organization functional class, adapted from the New York Heart Association class for the pulmonary hypertension context, sorts the child from class one, with no limitation, to class four, with symptoms at rest. The functional class, the six-minute walk distance in the older child, and the cardiopulmonary exercise test together quantify the severity and track the response to therapy. [5] [4]
The general paediatrician synthesises the findings into a one-line problem representation — for example, "an eight-year-old with exertional syncope, a loud second heart sound, and a right ventricular heave" — and that representation drives the urgent echocardiogram and the referral to the pulmonary hypertension service. The recognition is the generalist's job, and the definitive diagnosis and the therapy belong to the specialist team. [1] [6]
Investigations
Echocardiography is the first-line investigation, and it does three things at once. It estimates the pulmonary pressure from the tricuspid regurgitation velocity, it assesses the right ventricle for hypertrophy, dilation, and dysfunction, and it excludes or defines the congenital heart disease that may be driving the pressure. The septal geometry, the right ventricular size, and the pericardial effusion are the echocardiographic markers of severity, and they enter the risk stratification. [11] [1]
Cardiac catheterisation is the gold standard and is performed in every child before targeted therapy is started. It measures the mean pulmonary artery pressure, the wedge pressure, the cardiac output, and the indexed pulmonary vascular resistance, and it is the only investigation that confirms the diagnosis and separates the groups. The vasoreactivity test, performed with inhaled nitric oxide or oxygen, identifies the small subset of idiopathic and heritable patients who respond to high-dose calcium channel blockers, and a positive response is defined by a fall in the pressure and the resistance to near normal. [7] [1]
The bloods quantify the severity and the strain. The N-terminal pro-brain natriuretic peptide rises with right ventricular overload and is tracked over time, and the troponin and the renal and hepatic function gauge the end-organ impact of the low output. Iron studies are essential, because iron deficiency worsens the exercise tolerance and is common, and the genetic panel tests for bone morphogenetic protein receptor type 2 and the associated mutations in idiopathic and heritable disease. [4] [2]
The functional tests and the imaging complete the work-up. The six-minute walk distance and the cardiopulmonary exercise test quantify the exercise capacity in the older child, the ventilation-perfusion scan excludes chronic thromboembolic disease, the sleep study captures sleep-disordered breathing, and the cardiac magnetic resonance assesses the right ventricular volume and function. The chest radiograph shows cardiomegaly with prominent central pulmonary arteries and pruning of the peripheral vessels, and the electrocardiogram shows right ventricular hypertrophy and right axis deviation. [5] [12]
Management — Resuscitation
Resuscitation in the child with decompensated pulmonary hypertension targets the low cardiac output and the hypoxia, and the priority is to support the right ventricle while the definitive therapy is escalated. The child in right ventricular failure is managed in the paediatric intensive care unit, with oxygen, careful fluid management, and inotropic support to improve the contractility and the output. [4] [5]
Oxygen is a pulmonary vasodilator and is given to lower the resistance and improve the oxygenation, and it is particularly important in the group three lung disease where hypoxia drives the pressure. Fluid management is a fine balance, because the right ventricle on a stiff pulmonary bed is preload-sensitive, and over-resuscitation worsens the tricuspid regurgitation and the output. Inotropes such as milrinone and dobutamine improve the contractility and lower the afterload, and they are titrated to the output and the perfusion. [1] [4]
Inhaled nitric oxide is the selective pulmonary vasodilator of the intensive care unit, and it lowers the resistance without dropping the systemic pressure. It is used as a bridge in the decompensated child and as the agent for the vasoreactivity test at catheterisation. Extracorporeal membrane oxygenation is the rescue for the refractory low output, and it bridges the child to escalation of therapy or to transplantation. The general paediatrician's role is to recognise the decompensation and to arrange the urgent transfer to the pulmonary hypertension centre. [1] [7]
A caveat that examiners probe is the rhythm. Arrhythmia is both a consequence and a cause of decompensation, and the atrial arrhythmias of the dilated right atrium are poorly tolerated. The synchronised direct current cardioversion, the amiodarone, and the digoxin each have a role, and the intensivist and the electrophysiologist work together. The synchronised cardioversion is the move for the unstable rhythm, because the low output makes the medical option slow. [4] [5]
Management — Definitive & Stepwise
The definitive therapy is risk-stratified and built on combination treatment with drugs from the three pathways. The starting point is the risk assessment, which sorts the child into low, intermediate, or high risk using the functional class, the echocardiographic right ventricular function, the N-terminal pro-brain natriuretic peptide, and the haemodynamics. The risk level decides the intensity of the initial therapy and the urgency of the escalation. [4] [2]
The vasoreactive idiopathic and heritable subset is the one group treated with high-dose calcium channel blockers, and a positive response at catheterisation is required first, because the non-responders do poorly on these drugs. The non-vasoreactive child, which is the majority, is treated with combination therapy from the outset. The phosphodiesterase-5 inhibitor — sildenafil or tadalafil — protects the nitric oxide pathway, the endothelin receptor antagonist — bosentan, ambrisentan, or macitentan — blocks the excess endothelin, and these two oral agents are the usual initial combination for the low and intermediate risk child. [1] [10]
The prostacyclin pathway is added for the intermediate and high risk child. The inhaled iloprost, the subcutaneous or intravenous treprostinil, and the oral selexipag each deliver a prostacyclin effect, and the parenteral route is reserved for the high risk child with right ventricular failure. The sildenafil randomised trial in treatment-naive children demonstrated the benefit of the phosphodiesterase-5 inhibitor, and the modern practice combines the agents rather than using them in sequence, because the disease is driven by all three pathways at once. [10] [4]
The refractory child is escalated to the surgical options. The atrial septostomy creates a right-to-left pop-off that decompresses the right ventricle at the cost of a degree of cyanosis, and it is a bridge in the deteriorating child. The Potts shunt, an anastomosis between the left pulmonary artery and the descending aorta, is reserved for the child with suprasystemic idiopathic pulmonary arterial hypertension, and it unload the right ventricle by shunting to the lower body. Lung transplantation is the final option, and the timing of the referral is a judgement that balances the trajectory against the donor availability. [5] [2]
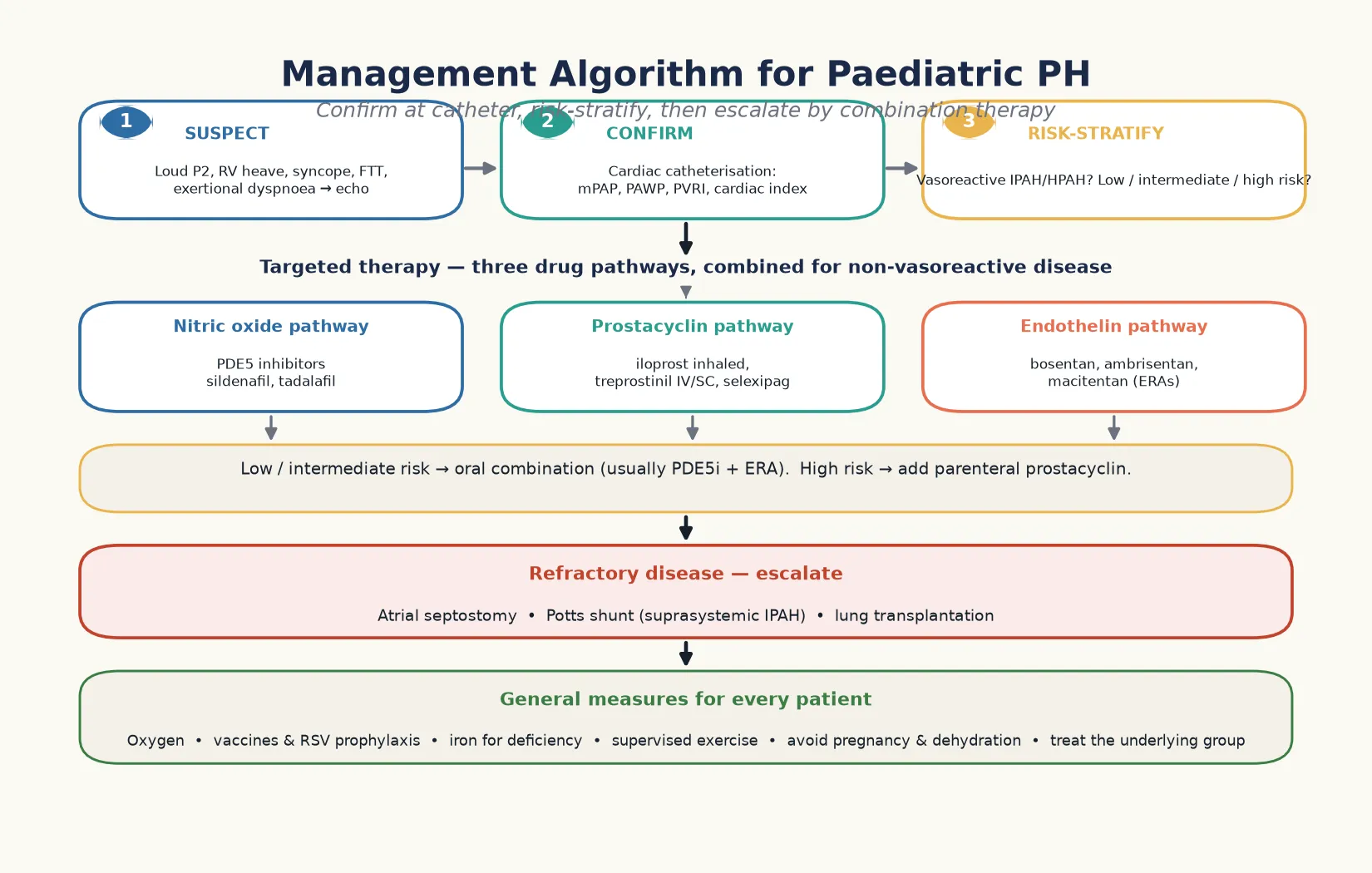
The risk-stratified escalation in five steps
Confirm the diagnosis and the group at cardiac catheterisation, including the wedge pressure and the vasoreactivity test.
Risk-stratify into low, intermediate, or high risk using the functional class, the echocardiographic right ventricular function, and the biomarkers.
Start oral combination therapy, usually a phosphodiesterase-5 inhibitor and an endothelin receptor antagonist, for the low and intermediate risk child.
Add a prostacyclin pathway agent, escalating to parenteral treprostinil for the high risk child with right ventricular failure.
Escalate to atrial septostomy, the Potts shunt, or lung transplantation for the refractory disease, and refer early for the transplant assessment.
Specific Subtypes & Scenarios
The congenital-heart-disease-associated form is the commonest paediatric subtype, and the management hinges on whether the shunt is still operable. A left-to-right shunt, such as a ventricular septal defect or an atrioventricular septal defect, raises the pulmonary flow and the pressure, and the resistance rises over time. Repair before the resistance crosses the threshold prevents the irreversible disease, and the threshold is an indexed resistance below a defined level, beyond which the operative risk is prohibitive and the shunt is left alone. The catheter, not the echo, decides the operability. [8] [5]
The Eisenmenger syndrome is the irreversible end of the unrepaired shunt, where the resistance has crossed the threshold and the shunt has reversed to right-to-left. The cyanosis, the clubbing, the secondary polycythaemia, and the exertional dyspnoea define the physiology, and the complications include haemoptysis, arrhythmia, heart failure, and the risks of pregnancy and non-cardiac surgery. The management is targeted therapy, anticoagulation in selected cases, and avoidance of pregnancy, and the modern drugs have improved the survival. The lesson is prevention — repair the shunt before the resistance crosses the threshold. [8] [2]
The bronchopulmonary-dysplasia-associated form is the dominant cause in the ex-preterm infant, and it presents as a failure to wean from oxygen, recurrent respiratory illness, and pulmonary hypertension on the echocardiogram. The mechanism is a combination of a reduced vascular bed, disordered angiogenesis, and chronic hypoxia, and the management is oxygen, nutrition, treatment of the lung disease, and the targeted therapy for the established pulmonary hypertension. The outcome is better than the idiopathic form, but the mortality is measurable, and the surveillance is lifelong. [9] [5]
The idiopathic and heritable form is the purest expression of the disease, and it is the one tested for vasoreactivity. The vasoreactive subset responds to high-dose calcium channel blockers and has a distinctly better prognosis, while the non-vasoreactive subset is treated with combination therapy. The genetic testing identifies the bone morphogenetic protein receptor type 2 and the associated mutations, and the family counselling follows, because the heritable form is autosomal dominant with variable penetrance. [2] [5]
Complications & Pitfalls
Right ventricular failure is the mode of death, and it is the complication that drives every escalation. The right ventricle hypertrophies to compensate, then dilates and fails as the afterload exceeds its capacity, and the signs are the rising jugular venous pressure, the hepatomegaly, the worsening tricuspid regurgitation, and the falling output. The early recognition of the failing right ventricle, before the frank cardiogenic shock, is the point at which the parenteral prostacyclin and the surgical options enter. [5] [4]
The arrhythmia of the dilated right atrium and right ventricle is both a consequence and a cause of decompensation. The atrial flutter and the atrial fibrillation are poorly tolerated, because the atrial contribution to the filling of a stiff right ventricle is significant, and the loss of the atrial kick drops the output. The management is rhythm control, with the amiodarone and the cardioversion, and the catheter ablation in selected cases. The intensivist and the electrophysiologist work together, because the rhythm and the output are inseparable. [4] [5]
The haemoptysis of the Eisenmenger syndrome is a feared complication, because it can be massive and life-threatening. The mechanism is the rupture of the hypertrophied bronchial arteries, and the management is conservative — avoiding anticoagulation in the acute bleed, treating the secondary polycythaemia, and the targeted therapy for the underlying disease. The non-cardiac surgery and the pregnancy in the Eisenmenger patient carry a high mortality, and the counselling is part of the lifelong care. [8] [2]
The diagnostic pitfalls share a common root: mistaking the early symptoms for deconditioning or asthma, and delaying the echocardiogram. A second pitfall is treating without measuring the wedge pressure, which harms the child with unrecognised left heart disease. A third is failing to risk-stratify, treating every child with a single oral agent when the high-risk child needs parenteral prostacyclin. The safeguard is the structured pathway — suspect, scan, catheterise, classify, escalate — and the early referral to the pulmonary hypertension service. [1] [7]
Prognosis & Disposition
The prognosis has improved markedly with the modern combination therapy, but the idiopathic form remains a life-limiting disease. The risk stratification sorts the child into low, intermediate, and high risk, and the high-risk child has a measurable one-year mortality that drives the escalation to parenteral prostacyclin and the transplantation referral. The functional class, the echocardiographic right ventricular function, the N-terminal pro-brain natriuretic peptide, and the haemodynamics together define the risk, and the risk is reassessed at every clinic visit. [4] [5]
The congenital-heart-disease-associated form has a better prognosis than the idiopathic form when the shunt is repaired in time, and the bronchopulmonary-dysplasia-associated form has an intermediate outlook. The Eisenmenger syndrome, once a disease with a poor prognosis, has improved with the targeted therapy, but it remains a condition with significant mortality from haemoptysis, arrhythmia, and heart failure. The heritable form behaves like the idiopathic form, and the vasoreactive subset has the best prognosis of all. [8] [2]
For the child who responds to therapy, the quality of life returns, the exercise tolerance improves, and the functional class falls. The therapy is lifelong, and the adherence is the determinant of the long-term outcome, because the disease progresses silently when the drugs are stopped. The transition to the adult pulmonary hypertension service happens in the late teens, and the structured transition protects the young person from being lost to follow-up at the point of greatest risk. [5] [12]
The general paediatrician owns the recognition and the referral, and the disposition is shared, structured care. The paediatric cardiology and the pulmonary hypertension service drive the diagnosis, the catheterisation, and the therapy. The intensive care service manages the decompensation, and the transplant service holds the final option. A named coordinator prevents the fragmentation that is the enemy of a lifelong plan, and the structured transition to the adult service protects the long-term outcome. [1] [4]
Special Populations
Pulmonary hypertension interacts with the child's genetic, social, and developmental context, and the same therapy behaves differently across populations. Access to a pulmonary hypertension service, to the expensive targeted drugs, and to the transplantation programme are the determinants of the outcome, and a plan that is clinically correct but unattainable for a family is no plan at all. [1] [5]
Indigenous children in Australia and Aotearoa New Zealand may face later presentation through reduced access to the echocardiogram and the specialist referral, and the congenital heart disease and the rheumatic heart disease that drive the pulmonary pressure may be more advanced at diagnosis. The retrieval service and the telehealth extend the diagnostic and the follow-up net into the remote communities, and the cultural safety of the counselling shapes the engagement. A low threshold to echocardiogram the child with a loud second heart sound, in any setting, is the safeguard. [12] [1]
The Down syndrome child carries a striking burden of pulmonary hypertension through several routes, and the screening is part of the routine care. The unrepaired atrioventricular septal defect, the developmental lung hypoplasia, the upper airway obstruction, and the persistent pulmonary hypertension of the newborn each contribute, and the cardiovascular assessment at every clinic visit is the safeguard. The migrant and the refugee family may arrive with an unrepaired shunt and no history, and the arrival health check includes the cardiovascular examination and the echocardiogram for any cyanosed or failing child. [9] [8]
Socioeconomic disadvantage shapes the access to the targeted drugs, the transport to the specialist centre, and the adherence to the lifelong therapy. The limiting step is often the cost and the logistics rather than the clinical decision, and the coordinator and the social work support bridge the gap. The young person is most often lost to follow-up at the transition to the adult service, and the targeted support at that transition protects the long-term outcome. The equity of the access is the measure of the quality of the service. [4] [5]
Evidence, Guidelines & Regional Differences
The evidence base rests on the American Heart Association and American Thoracic Society paediatric guideline, which is the operational standard for the diagnosis and the management of the paediatric disease, and the European Paediatric Pulmonary Vascular Disease Network consensus, which updated the risk-stratified approach and the combination therapy in 2019. These two documents frame the modern practice, and the fellowship candidate carries their principles. [1] [4]
The Rosenzweig 2019 European Respiratory Journal update and the Simonneau 2019 sixth World Symposium haemodynamic definitions refined the classification and the thresholds, and the Ivy 2013 Journal of the American College of Cardiology review remains a single-source reference for the paediatric disease. The Tracking Outcomes and Practice in Paediatric Pulmonary Hypertension registry papers provide the epidemiology and the catheterisation safety data that ground the paediatric numbers. [2] [3]
The sildenafil randomised trial in treatment-naive children demonstrated the benefit of the phosphodiesterase-5 inhibitor, and the Arvanitaki 2022 Eisenmenger review frames the management of the irreversible shunt. The 2015 European Society of Cardiology guideline, though written largely for adults, is endorsed by the Association for European Paediatric and Congenital Cardiology and provides the haemodynamic framework. The del Cerro bronchopulmonary-dysplasia study grounds the management of the ex-preterm form, and the Lammers echocardiography guide standardises the non-invasive assessment. [10] [11]
Where the evidence is weak, a fellowship answer says so honestly. The optimal timing of the escalation to parenteral prostacyclin is still being refined, and the role of the Potts shunt is accumulating evidence in the suprasystemic idiopathic form. The long-term outcome of the combination therapy in the young child, and the timing of the transplantation referral, are the active questions. The paediatric drug-dosing and the safety data lag the adult evidence, and much of the paediatric practice is extrapolated. Naming these uncertainties is a mark of intellectual honesty that examiners reward. [4] [2]
In Australia and Aotearoa New Zealand, paediatric pulmonary hypertension is managed according to the American Heart Association and American Thoracic Society guideline and the European consensus, with the specialist services concentrated in the major paediatric cardiac centres. The diagnosis and the therapy are delivered through a multidisciplinary pulmonary hypertension service, the targeted drugs are accessed through the special access schemes, and the retrieval service supports the remote and rural child. The transition to the adult service happens in the late teens, and the Cardiac Society of Australia and New Zealand provides the operational standard. [1] [4]
Exam Pearls
A fellowship candidate answering on pulmonary hypertension in children should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for, and the traps are where easy marks are lost. [1] [2]
Anchor one: define by the catheter, not the echo. Pulmonary hypertension is a mean pulmonary artery pressure above twenty at catheterisation, with pulmonary arterial hypertension requiring a raised resistance and a normal wedge. The echo estimates the pressure, but the catheter confirms the diagnosis and separates the groups. [3] [7]
Anchor two: classify by the WHO group. Group one is pulmonary arterial hypertension, including the idiopathic, heritable, and congenital-heart-disease-associated forms; group two is left heart disease; group three is lung disease; group four is thromboembolic; group five is multifactorial. The group decides the cause and the therapy. [3] [9]
Anchor three: the right ventricle decides survival. Right ventricular failure is the mode of death, so every management decision protects the right ventricle, not just the pressure number. A child in right ventricular failure is escalated to parenteral prostacyclin and the surgical options. [5] [4]
Anchor four: treat by combination therapy across the three pathways. Phosphodiesterase-5 inhibitors restore nitric oxide, endothelin receptor antagonists block the excess endothelin, and prostacyclin analogues replace what is lost. The high-risk child needs parenteral prostacyclin. [1] [10]
Anchor five: the Eisenmenger threshold is the must-not-miss judgement. Repair the shunt before the resistance crosses the threshold, because the Eisenmenger physiology is irreversible. The catheter, not the echo, decides operability. [8] [5]
The three traps to avoid are mistaking the early symptoms for deconditioning or asthma, treating without measuring the wedge pressure, and treating every child with a single oral agent when the high-risk child needs escalation. Exertional syncope is the red-flag symptom, the right ventricle decides the prognosis, and the catheter confirms the diagnosis — the high-yield facts a candidate holds. [1] [4]
References
- [1]Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, Hanna BD, Rosenzweig EB, Raj JU, Cornfield D, Stenmark KR, Steinhorn R, Thebaud B, Fineman JR, Kuehne T, Feinstein JA, Friedberg MK, Ewert P, Raj JU, Humpl T, American Heart Association, American Thoracic Society. Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation, 2015.PMID 26534956
- [2]Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Budts W, Channick RN, Delcroix M, Gatzoulis MA, Granton JT, Hirsch R, Ivy DD, Kulik TJ, Langleben D, Mathai SC, Mertens LL, Moledina S, Parker TA, Ranganathan S, Robin NH, Schulze-Neick I, Steinhorn RH, Van de Veerdonk MC, van Loon RLE, White RJ, Zijlstra WMP, Abman SH. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J, 2019.PMID 30545978
- [3]Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J, 2019.PMID 30545968
- [4]Hansmann G, Koestenberger M, Alastalo TP, Apitz C, Austin ED, Bonnet D, Budts W, D'Alto M, Gatzoulis MA, Hasan BS, Heng EL, Illina M, Kozlik-Feldmann R, Kuehne T, Lammers AE, Latus H, Moledina S, Muthurangu V, Moya Bonilla A, Ploegstra MJ, Rhode J, Schranz D, Warnecke G, Zijlstra WMP, Apitz C, Hansmann G. 2019 updated consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension: The European Pediatric Pulmonary Vascular Disease Network (EPPVDN), endorsed by AEPC, ESPR and ISHLT. J Heart Lung Transplant, 2019.PMID 31495407
- [5]Ivy DD, Abman SH, Barst RJ, Berger RMF, Bonnet D, Fleming TP, Haworth SG, Rosenzweig EB, Schulze Neick I, Steinhorn RH, Beghetti M. Pediatric pulmonary hypertension. J Am Coll Cardiol, 2013.PMID 24355636
- [6]Beghetti M, Berger RMF, Schulze-Neick I, Day RW, Kusic-Pajic A, Hislop AA, Ivy DD, Barst RJ, TOPP Registry Investigators. Diagnostic evaluation of paediatric pulmonary hypertension in current clinical practice. Eur Respir J, 2013.PMID 23563261
- [7]Beghetti M, Schulze-Neick I, Berger RMF, Day RW, Kusic-Pajic A, Ivy DD, Barst RJ, TOPP Registry Investigators. Haemodynamic characterisation and heart catheterisation complications in children with pulmonary hypertension: Insights from the Global TOPP Registry (tracking outcomes and practice in paediatric pulmonary hypertension). Int J Cardiol, 2016.PMID 26583838
- [8]Arvanitaki A, Gatzoulis MA, Opotowsky AR, Cordina R, Diller GP, Condliffe R, Law Y, Sheikh A, Dimopoulos K. Eisenmenger Syndrome: JACC State-of-the-Art Review. J Am Coll Cardiol, 2022.PMID 35331414
- [9]del Cerro MJ, Sabaté Rotés A, Cartón A, Deiros L, Bret M, Cordeiro M, Dos L, Garrido-Lestache E, Herraiz I, Abella R, Bialkowski J, Rueda F, Buba S, Grand B, Mendel B, Panos-Almansa JP, Sastre JA, Zunzunegui JL, Rios J, Abman SH, Berger RMF, Hislop AA, Ivy DD, Beghetti M, del Cerro MJ. Pulmonary hypertension in bronchopulmonary dysplasia: clinical findings, cardiovascular anomalies and outcomes. Pediatr Pulmonol, 2014.PMID 23788443
- [10]Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AN, Sastry BKS, Pulido T, Layton GR, Serdarevic-Pehar M, Kramer W. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation, 2012.PMID 22128226
- [11]Lammers AE, Apitz C, Michel-Behnke I, Daubeney P, Delhaas T, Diller GP, Gorenflo M, Hansmann G, Ilina M, Kegev R, Kozlik-Feldmann R, Kumlin M, Mertens L, Moledina S, Olma B, Okoromah C, Paleczek M, Rhode J, Schranz D, Tikitlis T, Wittelsbürger M, Zartner D, Apitz C, Lammers AE. A guide to echocardiographic assessment in children and adolescents with pulmonary hypertension. Cardiovasc Diagn Ther, 2021.PMID 34527541
- [12]Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, Aboyans V, Vaz Carneiro A, Achenbach S, Agewall S, Allanore Y, Asteggiano R, Paolo Badano L, Albert Barberà J, Bouvaist H, Bueno H, Byrne RA, Carerj S, Castro G, Erol Ç, Falk V, Funck-Brentano C, Gorenflo M, Granton J, Iung B, Kiely DG, Kirchhof P, Kjellström B, Landmesser U, Lekakis J, Lionis C, Lip GY, Orfanos SE, Park KM, Perrone-Filardi P, Ponikowski P, Revel MP, Rigau D, Rosenkranz S, Völler H, Luis Zamorano J, ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J, 2016.PMID 26320113