Paeds · cardiology
Tetralogy of Fallot
Also known as TOF · Fallot's tetralogy · Cyanotic congenital heart disease - tetralogy of Fallot
Fellowship guide to tetralogy of Fallot in children: the four features that share one embryologic fault, the severity spectrum from pink TOF to pulmonary atresia, the hypercyanotic tet spell and how to break it, the echo-to-MRI diagnostic strategy, primary repair versus staged palliation, the late burden of pulmonary regurgitation and sudden cardiac death, and the ANZ, AHA/ACC and ESC guideline positions.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the four-month-old brought in crying and blue after a feed, knees drawn up to the chest, who looks a little better when you hold her that way. Her mother has noticed her going duskier with feeds for weeks, and today she is saturating 72 per cent in air with a loud systolic murmur at the left upper sternal border. That child carries the whole story of tetralogy of Fallot — a cyanotic congenital heart defect whose severity swings with the degree of outflow obstruction, and whose most dangerous moment before surgery is the paroxysm you are now watching. [1]
Tetralogy of Fallot is the commonest cyanotic congenital heart lesion presenting beyond the neonatal period, with an incidence of roughly three to four per ten thousand live births. The four classical features — ventricular septal defect, right ventricular outflow tract obstruction, overriding aorta and right ventricular hypertrophy — are not four separate problems but the downstream consequences of a single malformation, anterocephalad deviation of the infundibular septum. This is the single most important concept for the viva: name the unifying embryologic fault and the four features follow. [7] [8]
The clinical importance of TOF rests on three facts. It is surgically curable, with operative mortality now under one to two per cent in modern centres, so the goal is recognition and safe transfer to surgery. It is the archetype of a duct-independent cyanotic lesion that nonetheless declares itself over weeks as obstruction worsens. And it is never truly "done" after repair, because the late burden of pulmonary regurgitation, arrhythmia and sudden cardiac death means every repaired patient needs lifelong follow-up. [1] [2]
Classification
The most useful way to classify TOF is by severity of the right ventricular outflow tract obstruction, because that single variable governs the direction of shunting, the age at presentation, and the urgency of repair. A second axis is the anatomy of the pulmonary outflow — classic TOF, TOF with pulmonary atresia, and TOF with absent pulmonary valve behave very differently. [1] [7]
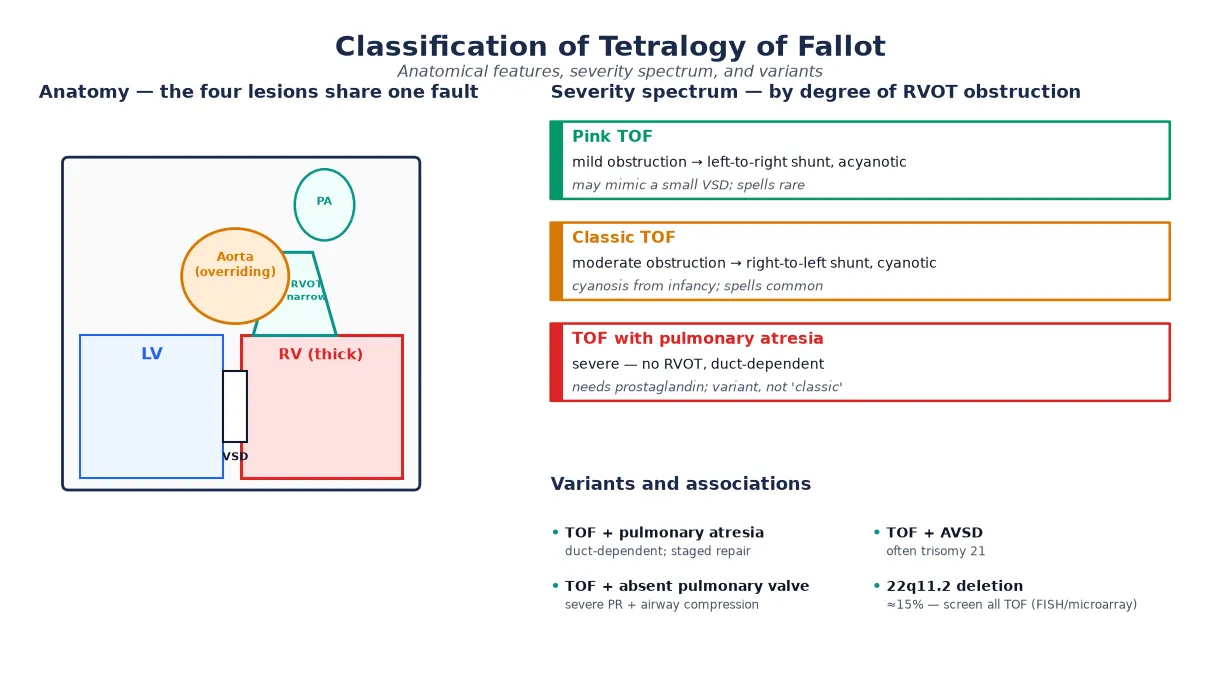
At the mild end is "pink" TOF, where the obstruction is slight and the shunt runs left-to-right, so the infant may be acyanotic and present only with a murmur and failure to thrive, sometimes confused with a small ventricular septal defect. In classic TOF, moderate obstruction produces a right-to-left shunt and cyanosis that emerges over the first weeks to months. At the severe end, TOF with pulmonary atresia has no continuity between the right ventricle and the pulmonary arteries, making the circulation duct-dependent from birth. [1] [8]
Pink TOF
mild obstruction
- Left-to-right shunt; acyanotic or trivial desaturation
- May mimic a VSD; spells uncommon
- Repair on the standard timeline
- Watch for worsening obstruction over time
Classic TOF
moderate obstruction
- Right-to-left shunt; cyanosis from infancy
- Hypercyanotic spells common
- Systolic ejection murmur at LUSB
- Repair around 3–6 months
TOF + pulmonary atresia
duct-dependent
- No RV-to-PA continuity; circulation duct-dependent
- Needs prostaglandin immediately
- Staged repair; may need RV-PA conduit
- Technically a distinct entity
TOF + absent pulmonary valve
variant
- Severe pulmonary regurgitation in utero
- Giant branch pulmonary arteries compress airways
- Respiratory failure may dominate
- Airway management drives the perioperative course
Two further variants deserve a viva mention. TOF with atrioventricular septal defect is uncommon and strongly associated with trisomy 21, with a more complex surgical repair. And the 22q11.2 deletion is found in around fifteen per cent of all TOF patients, so genetic testing is part of the workup of every child — it is not a variant of anatomy but a modifier of perioperative risk, calcium homeostasis, immune function and neurodevelopment. [5] [10]
Epidemiology & Risk Factors
TOF is uncommon in absolute terms but central in the paediatric curriculum because it is the commonest cyanotic lesion a general paediatrician will meet after the neonatal period. The incidence is around three to four per ten thousand live births, and it accounts for roughly seven to ten per cent of all congenital heart disease. There is a slight male predominance. Most cases are sporadic, but a family history of congenital heart disease and certain syndromes raise the background risk. [1]
The most important risk factor a candidate must name is the 22q11.2 deletion (DiGeorge syndrome), present in about fifteen per cent of TOF and in a higher proportion of TOF with pulmonary atresia. Other recognised associations are trisomy 21 (especially with the atrioventricular septal defect variant), Alagille syndrome (JAG1), and mutations in NKX2-5 and NOTCH1. Maternal diabetes, phenylketonuria, alcohol and certain anticonvulsants are recognised environmental contributors, and a right aortic arch is present in around twenty-five per cent. [1] [8]
The numbers that anchor your viva
Pathophysiology
The teaching model rests on a single embryologic fault that explains all four features and the clinical swings. During cardiac development, the infundibular (outlet) septum deviates anteriorly and superiorly, leaving a gap below it (the ventricular septal defect), narrowing the right ventricular outflow tract in front of it, and drawing the aortic root over the defect so the aorta appears to override. Right ventricular hypertrophy follows over weeks as the right ventricle pumps against the obstructed outflow. [7] [1]
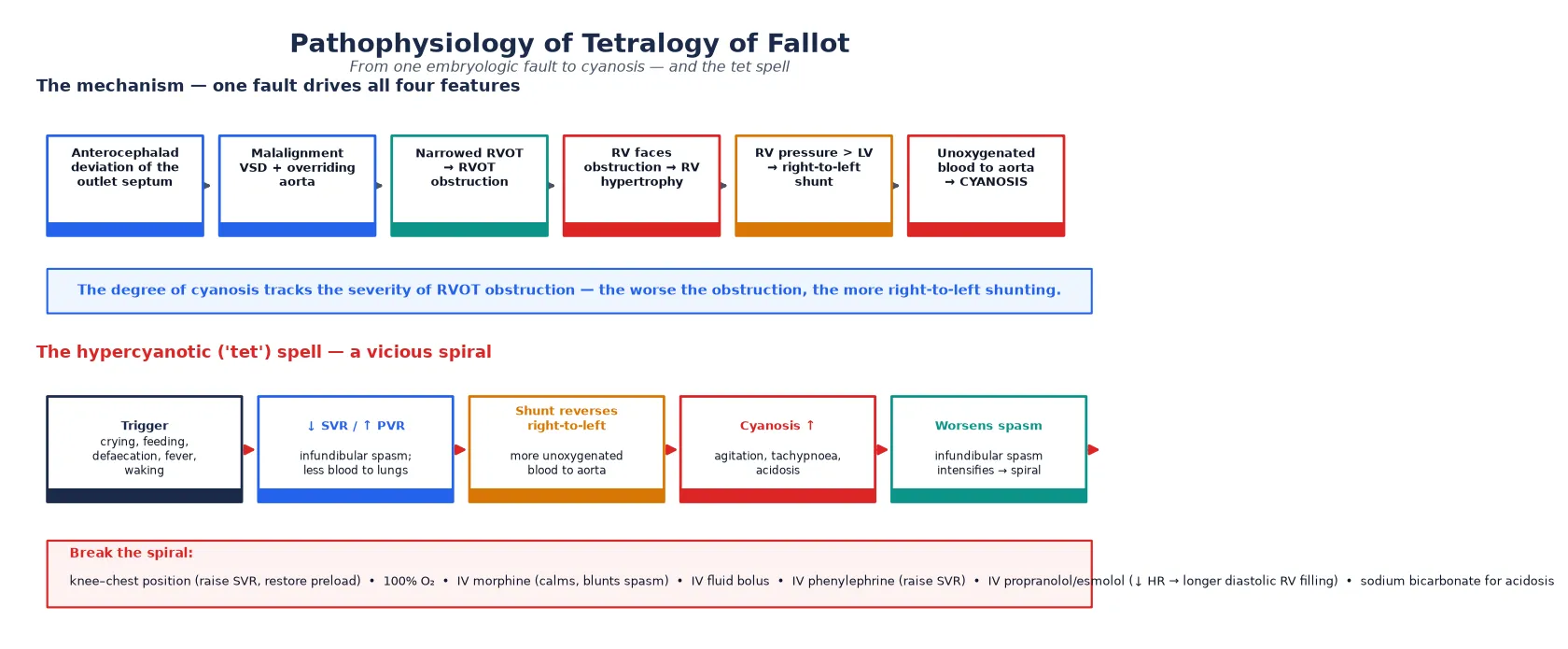
The degree of cyanosis tracks the severity of the right ventricular outflow tract obstruction. When the obstruction is mild, right ventricular pressure stays below systemic and the shunt runs left-to-right (pink TOF). As obstruction worsens, right ventricular pressure rises toward systemic, the shunt reverses to right-to-left, and unoxygenated blood enters the overriding aorta, producing cyanosis. This dynamic obstruction is why a child can be pink one hour and blue the next — the infundibular muscle is responsive to catecholamines and to physiological states. [1] [8]
The hypercyanotic "tet" spell is the clinical emergency that grows directly out of this physiology, and the examiner will expect a clean mechanism. A trigger — crying, feeding, defaecation, fever or waking — lowers systemic vascular resistance or raises pulmonary vascular resistance, and infundibular spasm narrows the outflow further. More blood is shunted right-to-left across the defect, cyanosis deepens, the child becomes agitated and tachypnoeic, acidosis develops, and the acidosis intensifies the infundibular spasm in a vicious spiral that can end in collapse and death if not broken. [1] [13]
[1]Clinical Presentation
The presentation depends almost entirely on the severity of the outflow obstruction, which is why TOF declares itself at different ages and in different ways. A neonate with severe obstruction or pulmonary atresia is cyanotic from birth and duct-dependent. The more typical infant is well for the first weeks, then becomes progressively duskier with feeds and crying, and is found to be desaturating at a routine check. Older children show squatting after exertion, a compensatory manoeuvre that raises systemic vascular resistance and transiently reduces the right-to-left shunt. [1] [15]
On examination the cardinal finding is a harsh systolic ejection murmur at the left upper sternal border radiating to the lung fields, generated by the right ventricular outflow tract obstruction rather than the ventricular septal defect. The second heart sound is single, because the pulmonary component is inaudible through the obstructed outflow. Cyanosis and clubbing develop over months, the precordium is quiet (unlike the hyperdynamic VSD), and a right aortic arch may be suggested by an asymmetric brachial pulse or a right-sided apex beat on imaging. [1] [8]
The hypercyanotic spell is the presentation that cannot be missed. The child, typically between two months and two years, becomes suddenly irritable, breathes fast and deeply, and turns intensely cyanotic, sometimes with loss of tone or a brief seizure from cerebral hypoxia. Spells are most common in the morning, with feeds, or after prolonged crying, and they resolve with squatting or knee–chest positioning. Any infant with known TOF who acutely deteriorates is having a spell until proven otherwise. [13] [1]
| Clinical picture | What it implies | Act |
|---|
Differential Diagnosis
Build the differential in layers, starting with the other causes of cyanosis in infancy and then the lesions that mimic an isolated murmur. The neonatal cyanosis differential centres on the five "T" lesions — tetralogy, transposition of the great arteries, tricuspid atresia, total anomalous pulmonary venous return, and truncus arteriosus — plus pulmonary atresia with intact ventricular septum and Ebstein anomaly. The hyperoxia test and echocardiography separate them. [1]
The murmur of TOF must be distinguished from other causes of a systolic murmur in infancy. An isolated ventricular septal defect gives a pansystolic murmur at the lower left sternal border with a hyperdynamic precordium, whereas TOF has an ejection murmur at the upper left sternal border and a quiet precordium. Pulmonary stenosis gives an ejection murmur but with normal oxygen saturation and a split second sound. Atrial septal defect has a fixed wide split and a pulmonary flow murmur. [8] [1]
Cyanotic mimics
- Transposition of the great arteries (more cyanotic, less murmur)
- Tricuspid atresia (left-axis deviation on ECG)
- Total anomalous pulmonary venous return
- Truncus arteriosus (single great vessel)
- Pulmonary atresia with intact septum
Murmur mimics
- Isolated ventricular septal defect (pansystolic, hyperdynamic)
- Pulmonary valve stenosis (saturations normal, split S2)
- Atrial septal defect (fixed split S2)
- Innocent Still's murmur (vibratory, normal sats)
- Coarctation (asymmetric pulses, not cyanotic)
Spell mimics
- Breath-holding attack (precipitant, rapid full recovery)
- Seizure with cyanosis (post-ictal state)
- Sepsis or metabolic acidosis
- Anaphylaxis
- Foreign-body airway obstruction
The decisive test is echocardiography, which defines the anatomy in a single study and resolves nearly all of these alternatives. The hyperoxia test (giving 100 per cent oxygen and watching for a rise in paO₂) historically separated cardiac from pulmonary cyanosis, but it is unreliable in duct-dependent lesions and is no substitute for urgent imaging; if pulmonary atresia is possible, start prostaglandin first. [1]
Clinical & Bedside Assessment
Assessment runs in parallel with stabilisation, because a cyanotic infant can deteriorate quickly. Secure the airway, breathing and circulation, attach a pulse oximeter, obtain intravenous access, and take a focused history while you examine, treating any emerging spell as you go. If the lesion might be duct-dependent, start a prostaglandin infusion at the same time as the assessment rather than waiting for confirmation. [1]
The history targets the timing and triggers of cyanosis (feeding, crying, morning), the frequency and severity of spells, failure to thrive, and a family history of congenital heart disease or a syndrome. Ask specifically about squatting in older children, which is virtually pathognomonic of unrepaired TOF. Take a careful perinatal and maternal history (diabetes, teratogens, infections) and screen for syndromic features that point to 22q11.2 or trisomy 21. [2] [10]
[1] [13]Examination looks for the degree of cyanosis and clubbing, the character of the precordium (quiet in TOF), the murmur described above, the second heart sound (single), the pulses in all four limbs (to detect a right aortic arch or coarctation), and the growth and dysmorphic features that suggest a syndrome. A careful, documented baseline examination is essential for tracking response to surgery and for the long-term surveillance that follows. [8] [1]
Investigations
Echocardiography is the cornerstone and is usually the only imaging needed to make the diagnosis and plan surgery. It demonstrates the malalignment ventricular septal defect, the overriding aorta, the site and severity of the right ventricular outflow tract obstruction, the size of the pulmonary arteries, and any associated lesions such as a right aortic arch or an atrial or ventricular septal defect. Colour and spectral Doppler estimate the gradient and the direction of shunting. [1] [2]
The chest X-ray gives the classic teaching sign of a "boot-shaped" heart, produced by the upturned apex of the hypertrophied right ventricle, with reduced pulmonary vascular markings and sometimes a right aortic arch. The electrocardiogram shows right-axis deviation and right ventricular hypertrophy with a dominant R in V1. These tests support the diagnosis but are not definitive — the echo is. [1] [8]
After repair, the cardiac magnetic resonance imaging study becomes the key investigation, because it quantifies right ventricular volumes and pulmonary regurgitation, the two variables that drive the timing of pulmonary valve replacement. Cardiac MRI also detects branch pulmonary artery stenosis, residual ventricular septal defect, and right ventricular outflow tract aneurysms. Cardiac catheterisation is reserved for haemodynamic questions, pre-PVR assessment, and transcatheter interventions. [6] [9]
The standard diagnostic workup
Pulse oximetry in all four limbs; full examination; four-limb blood pressures.
Echocardiography — diagnostic: defines the VSD, overriding aorta, RVOT obstruction and pulmonary arteries.
Chest X-ray (boot-shaped heart, pulmonary markings, aortic arch) and ECG (right-axis deviation, RVH).
Genetic testing for 22q11.2 deletion (FISH or chromosomal microarray) in every child.
Prenatal or fetal echocardiography when CHD is suspected on obstetric ultrasound.
After repair: serial cardiac MRI for right ventricular volumes and pulmonary regurgitation; ECG for QRS duration.
Prenatal diagnosis is increasingly important. A fetal echocardiogram performed for an abnormal obstetric anomaly scan, a family history, or a known syndrome can diagnose TOF from the second trimester, allowing planned delivery at a cardiac centre and early counselling of the family; series show good correlation between prenatal and postnatal anatomy. [12] [2]
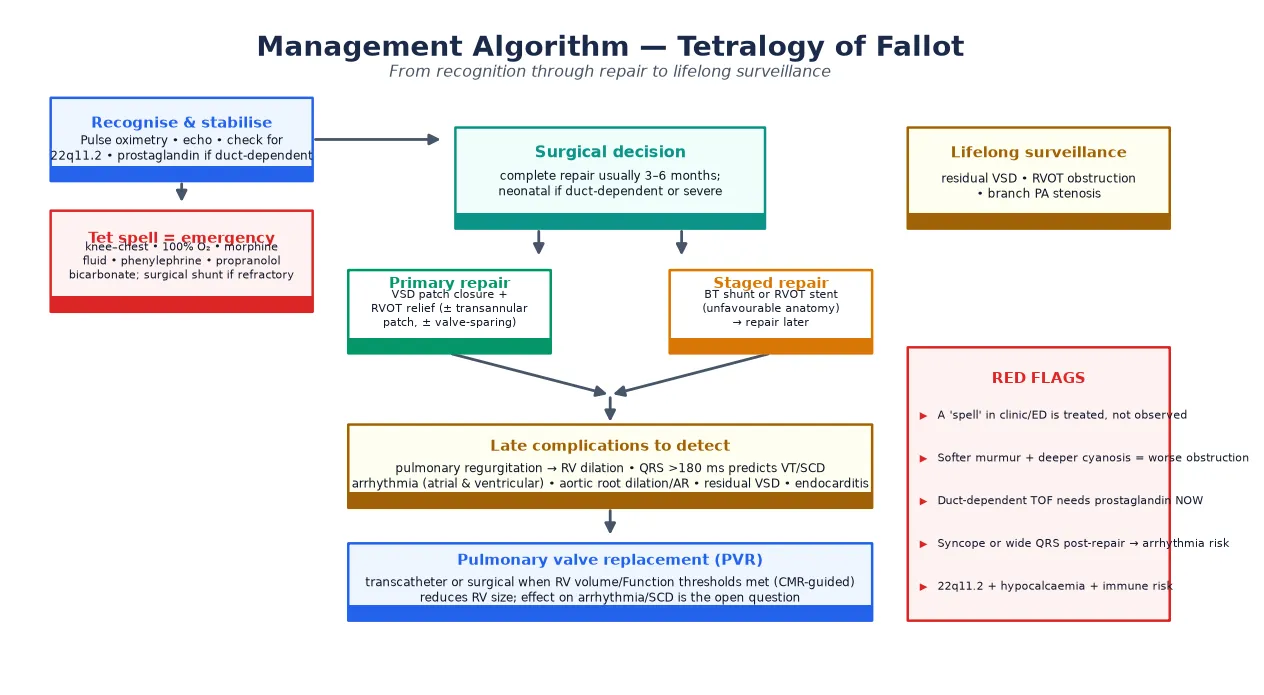
Management — Resuscitation
Resuscitation has two distinct scenarios: the hypercyanotic spell in the unrepaired infant, and the duct-dependent circulation in pulmonary atresia. Both are emergencies that precede definitive surgery and both reward prompt, protocol-driven action. Start with airway, breathing and circulation, give high-flow oxygen, establish intravenous access, and treat the spell or the duct-dependence before arranging transfer to a cardiac centre. [1]
For a hypercyanotic spell, place the child in the knee–chest position (which raises systemic vascular resistance and restores venous return to the right heart), give 100 per cent oxygen, and administer intravenous morphine at 0.1 mg/kg to calm the child and blunt infundibular spasm. Give an intravenous fluid bolus of 10 mL/kg isotonic saline to increase preload. If the spell continues, give intravenous phenylephrine at 5 micrograms/kg to raise systemic vascular resistance and force blood back across the outflow tract, or an intravenous beta-blocker such as propranolol or esmolol to slow the heart and allow longer diastolic filling of the right ventricle. Intranasal midazolam is a useful alternative when intravenous access is difficult. [1] [13]
SPELL-BREAK
For duct-dependent pulmonary atresia, start an intravenous infusion of prostaglandin E1 (alprostadil) at 0.01–0.05 micrograms/kg/minute immediately on suspicion, before the echo confirms the anatomy, because the duct is the only source of pulmonary blood flow. Prostaglandin apnoea, fever and hypotension are expected side effects, so plan for intubation if high doses are needed and transfer to a neonatal cardiac centre. A spell that does not respond to medical therapy needs an urgent surgical systemic-to-pulmonary shunt (modified Blalock–Taussig) or a transcatheter right ventricular outflow tract stent as a bridge to definitive repair. [14] [1]
Management — Definitive & Stepwise
Definitive management is surgical repair, and the modern standard is complete primary repair in infancy rather than the staged palliation of earlier eras. Complete repair closes the ventricular septal defect with a patch and relieves the right ventricular outflow tract obstruction by resection of infundibular muscle, with a transannular patch or a valve-sparing approach depending on the anatomy. The timing is typically three to six months of age for elective cases, with neonatal repair reserved for duct-dependent lesions or severe presentation. [1] [15]
Staged repair is reserved for infants with unfavourable anatomy — very small pulmonary arteries, severe outflow obstruction, or major aortopulmonary collaterals in pulmonary atresia. The first stage is a systemic-to-pulmonary shunt (modified Blalock–Taussig) or, increasingly, a transcatheter right ventricular outflow tract stent, which restores pulmonary blood flow and promotes pulmonary artery growth before a later complete repair. Stenting has emerged as a durable bridge in centres that cannot offer safe neonatal repair and in low-resource settings. [14] [2]
The technical choice at repair — transannular patch versus valve-sparing — has lifelong consequences and is a favoured viva topic. A transannular patch reliably relieves the obstruction but leaves the patient with free pulmonary regurgitation, which drives late right ventricular dilation, arrhythmia and the eventual need for pulmonary valve replacement. A valve-sparing or commisurotomy approach preserves pulmonary valve competence at the cost of a higher residual gradient, and is pursued where the native valve will accommodate it. The pendulum has swung toward valve-sparing to defer or avoid the burden of pulmonary regurgitation. [2] [9]
After repair, every patient enters a lifelong surveillance programme. The goals are to detect and treat residual right ventricular outflow tract obstruction, branch pulmonary artery stenosis, residual ventricular septal defect, aortic root dilation with aortic regurgitation, and the central late problem of pulmonary regurgitation with right ventricular dilation. Pulmonary valve replacement — surgical or, increasingly, transcatheter (percutaneous pulmonary valve) — is timed by cardiac MRI thresholds for right ventricular end-diastolic volume, ejection fraction and symptoms. [9] [6]
The life arc of repaired tetralogy of Fallot
The role of pulmonary valve replacement in reducing arrhythmia and sudden death is the open question of the era. Replacing the valve reliably reduces right ventricular size, but its effect on ventricular tachycardia and sudden cardiac death is less clear, which is why risk stratification — QRS duration, ventriculotomy scars, syncope, and electrophysiological study — guides the implantable cardioverter defibrillator decision separately from the volume decision. [2] [11]
Specific Subtypes & Scenarios
TOF with pulmonary atresia is technically a separate entity and the most demanding variant. There is no continuity between the right ventricle and the pulmonary arteries, and pulmonary blood flow depends on the ductus arteriosus or on major aortopulmonary collateral arteries. Management is individualised: some infants achieve unifocalisation and repair, others need a right ventricle-to-pulmonary artery conduit or remain in a single-ventricle pathway. Start prostaglandin immediately and involve a specialised surgical team. [14] [1]
TOF with absent pulmonary valve is a rare variant in which severe pulmonary regurgitation in utero produces aneurysmally dilated branch pulmonary arteries that compress the airways. The presentation may be dominated by respiratory failure rather than cyanosis, and airway management — sometimes including ventilation and bronchoscopic assessment — drives the perioperative course alongside the cardiac repair. [1] [8]
The adult with unrepaired TOF is increasingly rare in high-income settings but common globally, presenting with severe cyanosis, clubbing, exercise intolerance, secondary erythrocytosis and a high risk of brain abscess from the right-to-left shunt. Repair is still offered up to adulthood with acceptable results, though the preoperative burden of hypoxaemia leaves residual neurodevelopmental and haematological consequences. [15] [2]
Complications & Pitfalls
The complications divide into early postoperative problems and the late burden that drives lifelong surveillance. Early complications include residual ventricular septal defect, residual right ventricular outflow tract obstruction, branch pulmonary artery stenosis, heart block, junctional ectopic tachycardia, and the residual effects of cardiopulmonary bypass. Most are managed in the immediate postoperative period in a cardiac intensive care unit. [1] [2]
The late complications are where the fellowship marks are won. Pulmonary regurgitation is the central late lesion after a transannular patch repair, causing progressive right ventricular dilation, reduced exercise tolerance, and a substrate for arrhythmia. Atrial and ventricular arrhythmia, including sustained ventricular tachycardia, is common in adults, and sudden cardiac death is the most feared event — a QRS duration over 180 milliseconds on the electrocardiogram is a validated predictor of sustained ventricular tachycardia and sudden death. [3] [4]
The late risks that drive surveillance
Aortic root dilation with aortic regurgitation, infective endocarditis, residual ventricular septal defect, and the need for reoperation (most often pulmonary valve replacement) round out the late complications. The common pitfalls are clinical: failing to recognise a tet spell and observing rather than treating; failing to start prostaglandin in a duct-dependent neonate while awaiting echo; assuming a repaired patient is "cured" and discharging them from follow-up; and missing 22q11.2 deletion, with its implications for calcium, immune function and anaesthesia. [2] [5]
[2] [11]Prognosis & Disposition
The prognosis of repaired tetralogy of Fallot is one of the great success stories of paediatric cardiology. Surgical mortality in modern centres is under one to two per cent, and thirty-year survival after repair exceeds ninety per cent — a transformation from a lesion that was uniformly fatal in the first half of the twentieth century. Most repaired patients lead active lives, complete education, and have families. [15] [2]
The caveat is that "repaired" is not "cured." Late mortality is driven by sudden cardiac death and heart failure, and a substantial minority need at least one reoperation, most often pulmonary valve replacement. The duration and intensity of follow-up is individualised by the anatomy, the surgical technique, the residual lesions, and the arrhythmia risk, but no repaired patient is discharged from cardiac surveillance. Quality of life is generally good but is influenced by exercise capacity, neurodevelopment, and the psychological burden of a chronic condition. [3] [11]
Disposition after initial surgery is home with the family and the local paediatrician, with serial cardiac review. The adolescent must be prepared for transition to an adult congenital heart disease service — a structured handover of care that prevents the well-described "lost to follow-up" gap in early adulthood, when many sudden deaths and reoperations occur. Counselling about contraception, pregnancy risk, and the heritability of congenital heart disease is part of that transition. [2] [4]
Special Populations
Children with 22q11.2 deletion deserve special attention because the deletion modifies the whole care pathway. Beyond the cardiac lesion, they may have hypocalcaemia (especially neonatal), immune dysfunction with thymic aplasia, palatal abnormalities, feeding and speech difficulties, and a characteristic neurodevelopmental and behavioural phenotype. Perioperative care must address calcium monitoring, irradiated blood products, and airway and palate issues, and the family needs genetic counselling and a long-term developmental plan. [5] [10]
Infants with trisomy 21 and TOF, particularly the atrioventricular septal defect variant, require a coordinated cardiac-surgical and general-paediatric approach, with attention to associated gastrointestinal, airway and developmental issues. Resource-limited and remote populations — including Indigenous, refugee and rural communities — may present late with untreated disease, so culturally safe assessment, prompt retrieval, and a low threshold for prostaglandin are part of standard care. [14] [10]
The transitioning adolescent and young adult is now a defined population in congenital cardiology. The risks of lost follow-up, unplanned pregnancy, recreational drug and alcohol use, and non-adherence with surveillance converge in this group, so structured transition programmes — education, self-management skills, and a warm handover to the adult congenital service — are now standard and are emphasised in the AHA/ACC and CSANZ guidance. [2] [4]
Evidence, Guidelines & Regional Differences
| Region | Key guideline | Repair strategy | Pulmonary valve replacement |
|---|
The two controversies a candidate should be able to discuss are the timing and technique of the primary repair — transannular patch versus valve-sparing, neonatal versus elective — and the role of pulmonary valve replacement in reducing arrhythmia and sudden death. Practice has moved toward valve-sparing to protect the pulmonary valve, and toward earlier, CMR-guided valve replacement to limit right ventricular damage, but the evidence that replacement reduces sudden cardiac death remains incomplete, which is why electrophysiological risk stratification runs in parallel with the volume decision. [2] [11]
Exam Pearls
References
- [1]Apitz C, Webb GD, Redington AN Tetralogy of Fallot. Lancet, 2009.PMID 19683809
- [2]Villafañe J, Feinstein JA, Jenkins KJ, Vincent RN, Walsh EP, et al. Hot topics in tetralogy of Fallot. J Am Coll Cardiol, 2013.PMID 24076489
- [3]Khairy P, Aboulhosn J, Gurvitz MZ, Opotowsky AR, Mongeon FP, et al. Arrhythmia burden in adults with surgically repaired tetralogy of Fallot: a multi-institutional study. Circulation, 2010.PMID 20713900
- [4]Valente AM, Gauvreau K, Assenza GE, Babu-Narayan SV, Schreier J, et al. Contemporary predictors of death and sustained ventricular tachycardia in patients with repaired tetralogy of Fallot enrolled in the INDICATOR cohort. Heart, 2014.PMID 24179163
- [5]Mercer-Rosa L, Pinto N, Yang W, Tanel R, Goldmuntz E 22q11.2 Deletion syndrome is associated with perioperative outcome in tetralogy of Fallot. J Thorac Cardiovasc Surg, 2013.PMID 23312975
- [6]Joshi A, Ghadimi Mahani M, Dorfman A, Balasubramanian S Cardiac MR Evaluation of Repaired Tetralogy of Fallot. Semin Roentgenol, 2020.PMID 32859345
- [7]Anderson RH, Sarwark A, Spicer DE, Backer CL Exercises in anatomy: tetralogy of Fallot. Multimedia Manual Cardiothorac Surg, 2014.PMID 25500768
- [8]Anderson RH, Weinberg PM The clinical anatomy of tetralogy of fallot. Cardiol Young, 2005.PMID 15934690
- [9]Geva T Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu, 2006.PMID 16638542
- [10]McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers, 2015.PMID 27189754
- [11]Maury P, Sacher F, Rollin A, Mondoly P, Duparc A, et al. Ventricular arrhythmias and sudden death in tetralogy of Fallot. Arch Cardiovasc Dis, 2017.PMID 28222965
- [12]Grzyb A, Koleśnik A, Bokiniec R, Szymkiewicz-Dangel J Tetralogy of Fallot in the fetus - from diagnosis to delivery. 18-year experience of a tertiary Fetal Cardiology Center. Kardiol Pol, 2022.PMID 35579022
- [13]Montero JV, Nieto EM, Vallejo IR, Montero SV Intranasal midazolam for the emergency management of hypercyanotic spells in tetralogy of Fallot. Pediatr Emerg Care, 2015.PMID 25831027
- [14]Sandoval JP, Chaturvedi RR, Benson L, Morgan G, Van Arsdell G, et al. Right Ventricular Outflow Tract Stenting in Tetralogy of Fallot Infants With Risk Factors for Early Primary Repair. Circ Cardiovasc Interv, 2016.PMID 27965298
- [15]Bacha EA, Scheule AM, Zurakowski D, Erickson LC, Hung J, et al. Long-term results after early primary repair of tetralogy of Fallot. J Thorac Cardiovasc Surg, 2001.PMID 11436049