Paeds · cardiology
Ventricular arrhythmias and sudden cardiac death
Also known as Ventricular tachycardia · VT · Ventricular fibrillation · VF · Sudden cardiac death · SCD in the young · Sudden arrhythmic death syndrome · SADS · Commotio cordis · Idiopathic ventricular fibrillation
Fellowship guide to ventricular arrhythmias and sudden cardiac death in children: distinguishing benign ventricular ectopy from sustained monomorphic and polymorphic VT and VF, the four cause families of SCD in the young (structurally normal heart, cardiomyopathy, congenital heart disease, acquired), the acute resuscitation of pulseless VT and VF with defibrillation and amiodarone, the synchronised cardioversion versus pharmacological termination of stable sustained VT, post-arrest targeted temperature management, ICD therapy and its paediatric-specific complications, the molecular autopsy and cascade screening, and the AHA/ACC/HRS and ESC guideline positions.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the fourteen-year-old boy who collapses without warning on a soccer pitch. He was sprinting for the ball, there was no contact, and he is now pulseless and apnoeic. A bystander begins CPR and the automated external defibrillator advises a shock. That child carries the whole story of ventricular arrhythmias and sudden cardiac death in the young: a previously apparently well child who has a clinically silent substrate — a cardiomyopathy, a channelopathy, or a coronary anomaly — and in whom an electrical storm of ventricular tachycardia or ventricular fibrillation strikes without prodrome, often during exertion, and produces cardiac arrest. Whether he survives depends entirely on what happens in the next three minutes, and on whether the underlying cause is subsequently identified and treated. [1] [3]
Ventricular arrhythmias are abnormal rhythms originating below the atrioventricular node in the His-Purkinje system or the ventricular myocardium. They span a spectrum from premature ventricular complexes (isolated ectopic beats that are usually benign in the structurally normal heart), through non-sustained ventricular tachycardia (three or more consecutive ventricular beats lasting less than thirty seconds), to sustained ventricular tachycardia (lasting more than thirty seconds or producing haemodynamic compromise) and ventricular fibrillation (chaotic, disorganised electrical activity producing no effective cardiac output). The clinical urgency escalates along this spectrum, and the management divides sharply at the presence or absence of a pulse. [2] [4]
Sudden cardiac death is defined as an unexpected death from a cardiac cause occurring within one hour of symptom onset (or unwitnessed, within twenty-four hours of last being seen alive), in a person without a prior condition that would appear fatal. In children and young adults, the annual incidence is approximately one to three per hundred thousand, which makes it rare in absolute terms but one of the leading medical causes of death in this age group — and because a substantial proportion of cases have a heritable substrate, every sudden death in a young person carries implications for the surviving relatives. [1]
Classification
The most useful way to classify ventricular arrhythmias is first by the rhythm itself (which determines the immediate resuscitation), and second by the underlying cause (which determines the long-term strategy). The rhythm classification runs from isolated ventricular ectopy through sustained monomorphic and polymorphic VT to ventricular fibrillation; the cause classification divides sudden cardiac death in the young into four families that together cover virtually every clinical scenario. [1] [2]
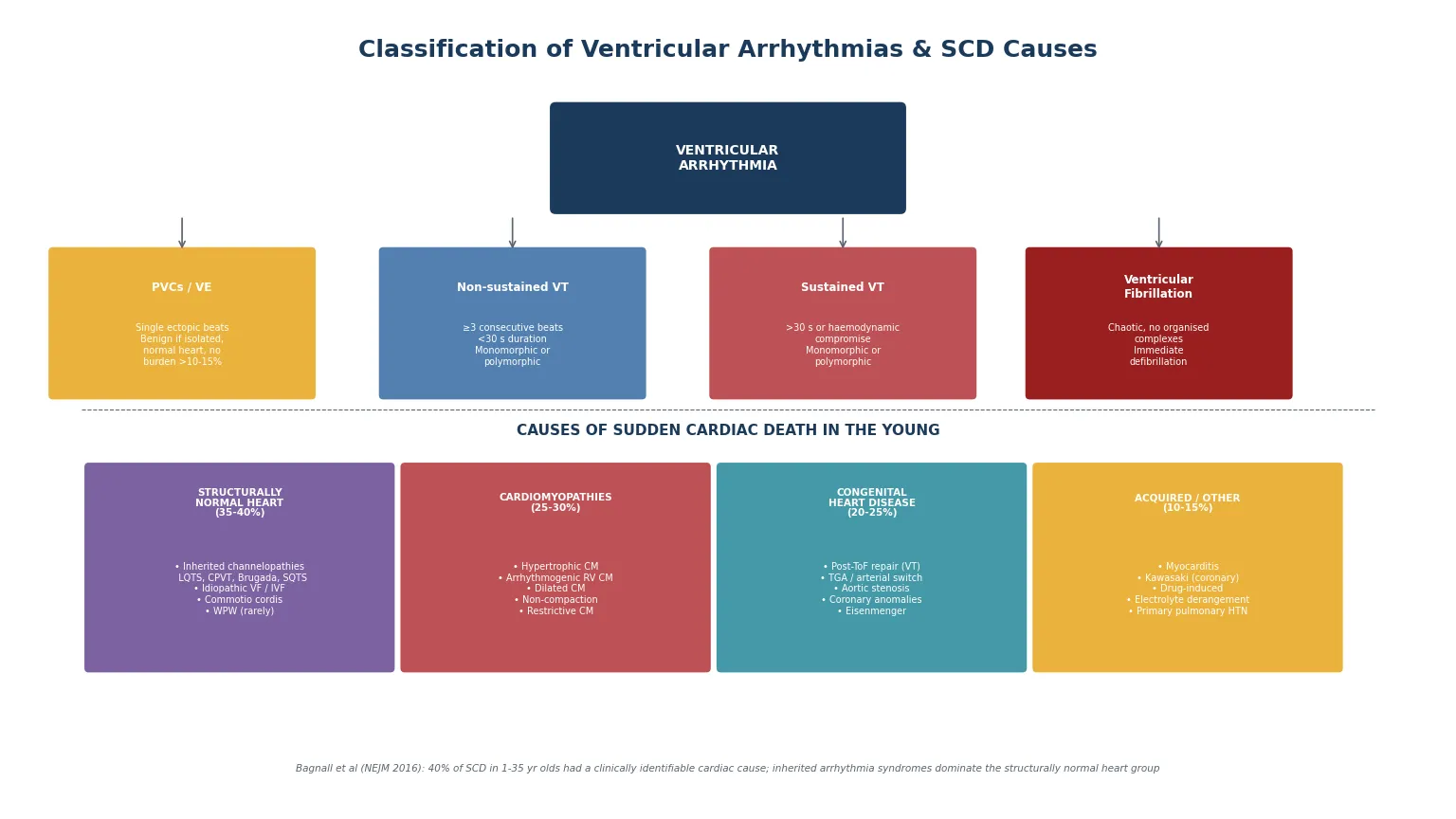
The rhythm classification is operationally critical. Premature ventricular complexes are identified by a wide QRS complex not preceded by a P wave; they are common in children and, in the structurally normal heart with a low burden (typically less than ten to fifteen per cent of total beats on ambulatory monitoring and absent couplets or runs), they are benign. Non-sustained VT — three or more consecutive ventricular beats at a rate above one hundred and twenty per minute, lasting under thirty seconds — warrants investigation but is often asymptomatic. Sustained VT (lasting more than thirty seconds or causing haemodynamic compromise) and ventricular fibrillation are emergencies. The further distinction is monomorphic versus polymorphic: monomorphic VT suggests a fixed anatomical substrate (a scar, an outflow-tract focus), while polymorphic VT suggests a dynamic trigger (ischaemia, QT prolongation with torsades, or catecholaminergic stress). [2] [4]
PVCs / Ventricular ectopy
benign if isolated
- Wide QRS, no preceding P wave
- Common in children; usually benign
- Burden <10-15% on Holter, no couplets or VT runs → reassurance
- Red flags: polymorphic, couplets, exercise-provoked, high burden → investigate
- Underlying structural disease or channelopathy must be excluded
Sustained monomorphic VT
fixed substrate
- Regular, uniform wide-QRS tachycardia >30 s
- Re-entry around scar: post-ToF repair, ARVC
- Automatic focus: RVOT VT, fascicular (Belhassen) VT
- Often amenable to catheter ablation if idiopathic
- Haemodynamic stability varies with rate and substrate
Polymorphic VT / Torsades
dynamic trigger
- Twisting QRS axis, varying morphology
- QT prolongation → torsades de pointes (LQTS, acquired)
- CPVT → bidirectional VT on exercise
- Ischaemia, electrolyte disturbance
- High risk of degeneration to VF
Ventricular fibrillation
arrest rhythm
- Chaotic, disorganised electrical activity
- No effective cardiac output
- Immediate defibrillation required
- May be primary (channelopathy, commotio cordis) or secondary (degeneration of VT)
- Survival depends on bystander CPR and early shock
The cause classification of sudden cardiac death divides into four families. The structurally normal heart group — approximately thirty-five to forty per cent of SCD in the young in the Bagnall cohort — comprises the inherited arrhythmia syndromes (long-QT syndrome, catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome, short-QT syndrome), idiopathic ventricular fibrillation, and commotio cordis. The cardiomyopathies — twenty-five to thirty per cent — are dominated by hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy, with dilated, restrictive, and non-compaction cardiomyopathy also contributing. Congenital heart disease — twenty to twenty-five per cent — is driven by repaired tetralogy of Fallot, transposition of the great arteries after atrial or arterial switch, aortic stenosis, and coronary artery anomalies. Acquired causes — ten to fifteen per cent — include myocarditis, Kawasaki disease with coronary artery aneurysms, drug-induced arrhythmia, and severe electrolyte disturbance. [1] [8]
Epidemiology & Risk Factors
The incidence of sudden cardiac death in children and young adults (ages one to thirty-five) is approximately one to three per hundred thousand per year, with higher rates in adolescence and the early twenties and in males. It is rare in absolute terms — a general paediatrician may not encounter a case in a career — but it is one of the leading medical causes of death in this age group, and the social impact of a young athlete dying on the field is disproportionate. The incidence is higher in populations with a high prevalence of hypertrophic cardiomyopathy (e.g., competitive athletes) and in families with known inherited cardiac conditions. [1] [3]
The prospective Australian study by Bagnall and colleagues, which examined all sudden cardiac deaths in one- to thirty-five-year-olds in Australia and New Zealand over three years, remains the definitive epidemiological study. It found that a clinically identifiable cardiac cause was present in forty per cent of cases, that the structurally normal heart group was dominated by inherited arrhythmia syndromes, and that a substantial proportion of deaths were potentially preventable if the underlying condition had been recognised earlier. The study underpins the recommendation that every unexplained syncope, seizure, or sudden death in a young person warrants a twelve-lead ECG and, where indicated, specialist evaluation. [1]
The numbers that anchor your viva
The most important risk stratification variables, in any child presenting with a ventricular arrhythmia, are the presence of structural heart disease (cardiomyopathy, congenital heart disease), the symptom history (syncope, aborted cardiac arrest), the family history of sudden death before age forty, and the resting twelve-lead ECG (QTc, repolarisation pattern, pre-excitation). A child who has already survived a cardiac arrest is at the highest risk of recurrence and mandates an ICD for secondary prevention. [3] [4]
Pathophysiology
The teaching model rests on three cellular mechanisms that produce ventricular arrhythmia — re-entry, enhanced automaticity, and triggered activity (early and delayed afterdepolarisations) — and on the principle that any sustained ventricular arrhythmia, if not terminated, degenerates into ventricular fibrillation and produces cardiac arrest. Understanding the mechanism guides both the acute termination and the long-term strategy, because each mechanism responds differently to antiarrhythmic drugs, ablation, and device therapy. [2] [8]
Re-entry is the commonest mechanism for sustained monomorphic VT in structural heart disease. A re-entrant circuit requires a unidirectional block, a slow conduction pathway, and a recovering tissue ahead of the advancing wavefront — conditions created by scar (post-surgical, post-myocarditis), by the fibro-fatty replacement of arrhythmogenic right ventricular cardiomyopathy, or by the disordered architecture of hypertrophic cardiomyopathy. The classic paediatric re-entry substrate is the post-operative tetralogy of Fallot patient, in whom the right ventricular outflow tract patch, the ventriculotomy scar, and the conal septum create anatomical isthmuses that support macro-re-entrant VT years after repair. [4]
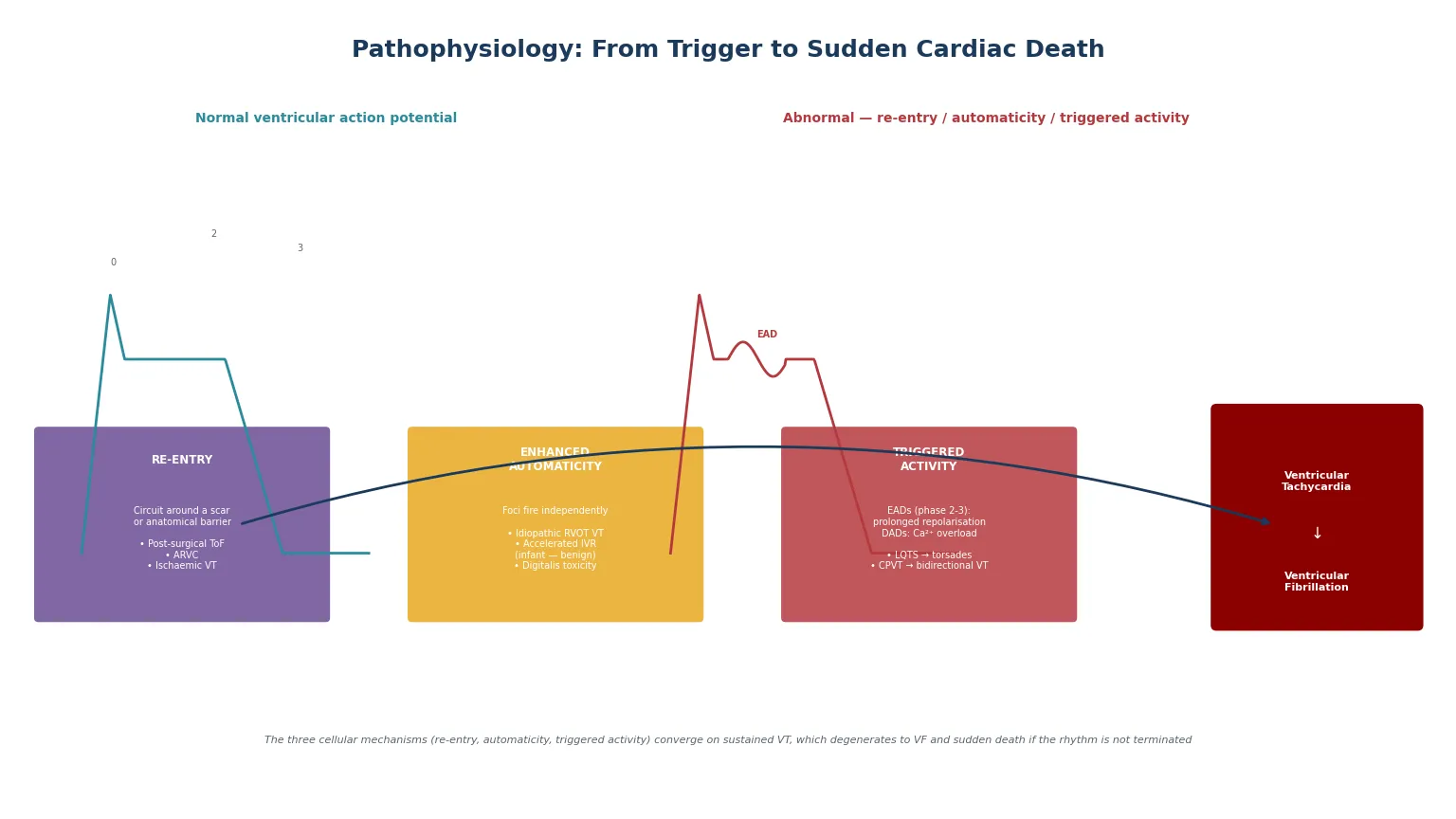
Enhanced automaticity describes a focus that fires independently of the surrounding myocardium. The classic paediatric automatic ventricular arrhythmias are right ventricular outflow tract VT (a focal, exercise-related, often adenosine-sensitive tachycardia that is generally benign and amenable to catheter ablation) and accelerated idioventricular rhythm in infants (a slightly faster-than-sinus rhythm with a wide QRS that is benign and usually resolves in the first year of life). [2] [10]
Triggered activity is driven by afterdepolarisations — secondary depolarising oscillations that occur during or after the action potential. Early afterdepolarisations (arising during phases two and three) are produced by prolonged repolarisation and underlie the torsades de pointes of long-QT syndrome. Delayed afterdepolarisations (arising after full repolarisation, in phase four) are produced by intracellular calcium overload and underlie the bidirectional VT of catecholaminergic polymorphic ventricular tachycardia and the arrhythmias of digitalis toxicity. Both mechanisms are adrenergically driven, which is why exercise and emotion are common triggers for channelopathy-related events. [8] [12]
[5] [6]Whatever the mechanism, a sustained ventricular arrhythmia produces haemodynamic collapse by two routes: loss of atrioventricular synchrony (the ventricles fill poorly without atrial contribution) and the rapid rate itself (diastolic filling time is too short for adequate preload). At very fast rates or with polymorphic rhythms, the ventricular contraction becomes ineffective and cardiac output ceases — ventricular fibrillation. The cerebral cortex tolerates only three to four minutes of no-flow before irreversible injury begins, which is why bystander CPR and early defibrillation are the single most important determinants of survival. [3] [7]
Clinical Presentation
The presentation of ventricular arrhythmia ranges from an incidental finding on a monitor or ambulatory ECG, through palpitations and presyncope, to syncope, seizure-like activity, and sudden cardiac death. The context of presentation — exertional versus resting, witnessed versus unwitnessed, with or without prodrome — is the single most important diagnostic clue. [1] [8]
The classic high-risk presentations are syncope during or immediately after exertion (suggesting hypertrophic cardiomyopathy, a coronary anomaly, CPVT, or long-QT type one), syncope triggered by emotion or auditory stimuli (suggesting CPVT or long-QT type two), and sudden collapse with no prodrome during sleep (suggesting long-QT type three or Brugada syndrome). Syncope during exertion is never vasovagal until proven otherwise — this is the cardinal rule that protects against the missed diagnosis of a life-threatening arrhythmia. Seizure-like movements during the arrhythmic event are common and frequently misdiagnosed as epilepsy, with children sometimes treated with anticonvulsants for years before the underlying arrhythmia is identified. [8] [12]
| Clinical picture | What it implies | Act |
|---|
Neonatal and infantile presentations include accelerated idioventricular rhythm (a benign, self-limiting rhythm in the first year of life that resolves spontaneously), ventricular ectopy (common, usually benign if isolated and the heart is structurally normal), and rare severe early-onset VT from calmodulin mutations or congenital long-QT syndrome presenting with two-to-one atrioventricular block. The post-operative congenital heart disease patient may present with VT years after repair, particularly after tetralogy of Fallot repair, which is why these patients undergo lifelong surveillance with ambulatory ECG and exercise testing. [2] [4]
Differential Diagnosis
Build the differential in two layers: first, the causes of a wide-QRS tachycardia on the ECG or monitor, and second, the causes of sudden collapse in a child. The distinction between ventricular tachycardia and supraventricular tachycardia with aberrancy is the classic pitfall, and the safe default in a haemodynamically compromised child with a regular wide-QRS tachycardia is to treat it as VT until proven otherwise. [3] [4]
A regular wide-QRS tachycardia (QRS duration above the age-specific upper limit, typically more than one hundred and twenty milliseconds in adolescents) is most commonly ventricular tachycardia in a child with structural heart disease, but supraventricular tachycardia with aberrancy, pre-excited tachycardia (atrioventricular re-entry tachycardia using an accessory pathway, as in Wolff-Parkinson-White syndrome with atrial fibrillation), and ventricular-paced rhythms must be excluded. The Brugada criteria for VT (atrioventricular dissociation, capture beats, fusion beats, concordance of precordial QRS) are less well validated in children, so the clinical context — structural heart disease, electrolyte disturbance, drug toxicity — is critical. [2] [10]
Wide-QRS tachycardia causes
- Ventricular tachycardia (the default in structural disease)
- SVT with aberrancy (bundle branch block)
- Pre-excited tachycardia (WPW with AF)
- Ventricular-paced rhythm
- Treat the unstable child as VT until proven otherwise
Collapse mimics
- Vasovagal syncope (prodrome, rapid recovery)
- Breath-holding attacks in toddlers
- Seizure / convulsive syncope
- Hypoglycaemia
- Anaphylaxis / choking / drowning
SCD cause families
- Structurally normal heart: LQTS, CPVT, Brugada, idiopathic VF, commotio cordis
- Cardiomyopathy: HCM, ARVC, DCM, non-compaction
- Congenital heart disease: post-ToF, coronary anomaly, aortic stenosis
- Acquired: myocarditis, Kawasaki, drugs, electrolytes
The causes of sudden collapse without an ECG in hand must be rapidly separated, because the resuscitation priorities differ. Anaphylaxis requires intramuscular adrenaline; choking or drowning requires airway management; hypoglycaemia requires glucose; but the child in cardiac arrest from VF or pulseless VT requires defibrillation within minutes. The single most important step is obtaining a rhythm strip as early as possible, ideally with the automated external defibrillator pads, and beginning high-quality CPR immediately. [3] [7]
Clinical & Bedside Assessment
Assessment of the child with a suspected ventricular arrhythmia runs from the history through the twelve-lead ECG, and in the acute event it is inseparable from the resuscitation. The general paediatrician must be able to recognise the dangerous rhythm on the monitor and initiate the shockable-rhythm loop without waiting for specialist arrival. [3] [4]
The history targets the circumstances of any preceding events: was there syncope, and if so was it exertional, emotional, nocturnal, or triggered by a specific stimulus? Was there a prodrome (suggesting vasovagal) or a sudden, unheralded collapse (suggesting arrhythmia)? Is there a family history of sudden death, syncope, seizures, deafness, or known cardiac disease in first-degree relatives? Is the child taking any medications (particularly QT-prolonging agents or stimulants)? In the congenital heart disease patient, what was the surgical history, and when was the last cardiology follow-up? [1] [8]
[3] [8]The physical examination in the stable child focuses on signs of structural heart disease (murmurs, the ejection systolic murmur and paradoxical splitting of hypertrophic cardiomyopathy, the findings of repaired tetralogy), features of syndromes associated with cardiomyopathy or channelopathy (Marfanoid habitus, features of Noonan or Williams syndrome), and signs of heart failure. In the acute event, the examination is the standard airway-breathing-circulation assessment of the collapsed child, with immediate rhythm interpretation. [2] [4]
Investigations
The diagnostic strategy in the stable child has four tiers: the resting twelve-lead ECG, the ambulatory and exercise ECG, the echocardiogram and cardiac imaging, and the genetic and metabolic evaluation. The aim is to identify the arrhythmia substrate and stratify risk, because the long-term strategy — ICD, ablation, disease-modifying therapy, or reassurance — depends entirely on the cause. [2] [4]
The resting twelve-lead ECG is the single most important investigation. Measure the QTc manually (Bazett correction, confirmed with Fridericia in children); assess for left ventricular hypertrophy and repolarisation abnormalities (suggesting cardiomyopathy); look for the epsilon waves and T-wave inversion in V1 to V3 of arrhythmogenic right ventricular cardiomyopathy; look for the coved ST elevation of Brugada syndrome; and look for pre-excitation (delta wave of Wolff-Parkinson-White). A normal resting ECG does not exclude a channelopathy — CPVT has a structurally normal heart and a normal resting ECG, and the exercise test is diagnostic. [8] [12]
The standard diagnostic workup after a ventricular arrhythmia
12-lead ECG with manual QTc measurement and assessment for hypertrophy, repolarisation abnormalities, epsilon waves, Brugada pattern, and pre-excitation.
Echocardiography to identify structural heart disease (cardiomyopathy, congenital heart disease, coronary anomalies, ventricular dysfunction).
Ambulatory ECG (Holter) for twenty-four to forty-eight hours to assess ectopic burden, couplets, non-sustained VT, and heart rate variability.
Exercise stress test (mandatory if CPVT suspected — normal resting ECG, bidirectional or polymorphic VT on exercise; also assesses QT adaptation in LQT1).
Cardiac MRI for tissue characterisation (late gadolinium enhancement in ARVC and HCM; ventricular volumes and function post-ToF repair).
Electrolyte panel (potassium, calcium, magnesium), toxicology screen, and thyroid function (hyperthyroidism can destabilise arrhythmia).
Genetic testing through an inherited cardiac conditions service (targeted or panel testing guided by the phenotype); molecular autopsy if the proband has died.
Cascade screening of all first-degree relatives with ECG and, if a mutation is found, targeted genetic testing.
The exercise stress test is the key investigation for catecholaminergic polymorphic ventricular tachycardia, in which the resting ECG is normal and the arrhythmia is provoked by adrenergic stress. The exercise test in CPVT reveals the characteristic bidirectional or polymorphic ventricular ectopy at higher heart rates, typically above one hundred and ten to one hundred and twenty beats per minute, progressing to non-sustained or sustained VT with increasing workload. Any child with exertional syncope and a normal resting ECG needs an exercise test before the arrhythmia is dismissed. [8] [12]
Cardiac MRI is increasingly central to the evaluation of the structurally abnormal heart, because late gadolinium enhancement identifies the fibrosis that underlies re-entrant VT in arrhythmogenic right ventricular cardiomyopathy, hypertrophic cardiomyopathy, and post-surgical congenital heart disease. Genetic testing — guided by the phenotype and performed through an inherited cardiac conditions service with pre- and post-test counselling — identifies the causative mutation in a substantial proportion of channelopathies and cardiomyopathies, and enables predictive (cascade) testing of relatives. [2] [4]
Management — Resuscitation
The acute event is the pulseless ventricular tachycardia or ventricular fibrillation arrest, and the management follows the shockable-rhythm loop of the paediatric advanced life support algorithm. The general paediatrician must be able to lead this resuscitation, because the outcome depends on the quality and speed of the early response. [3] [7]
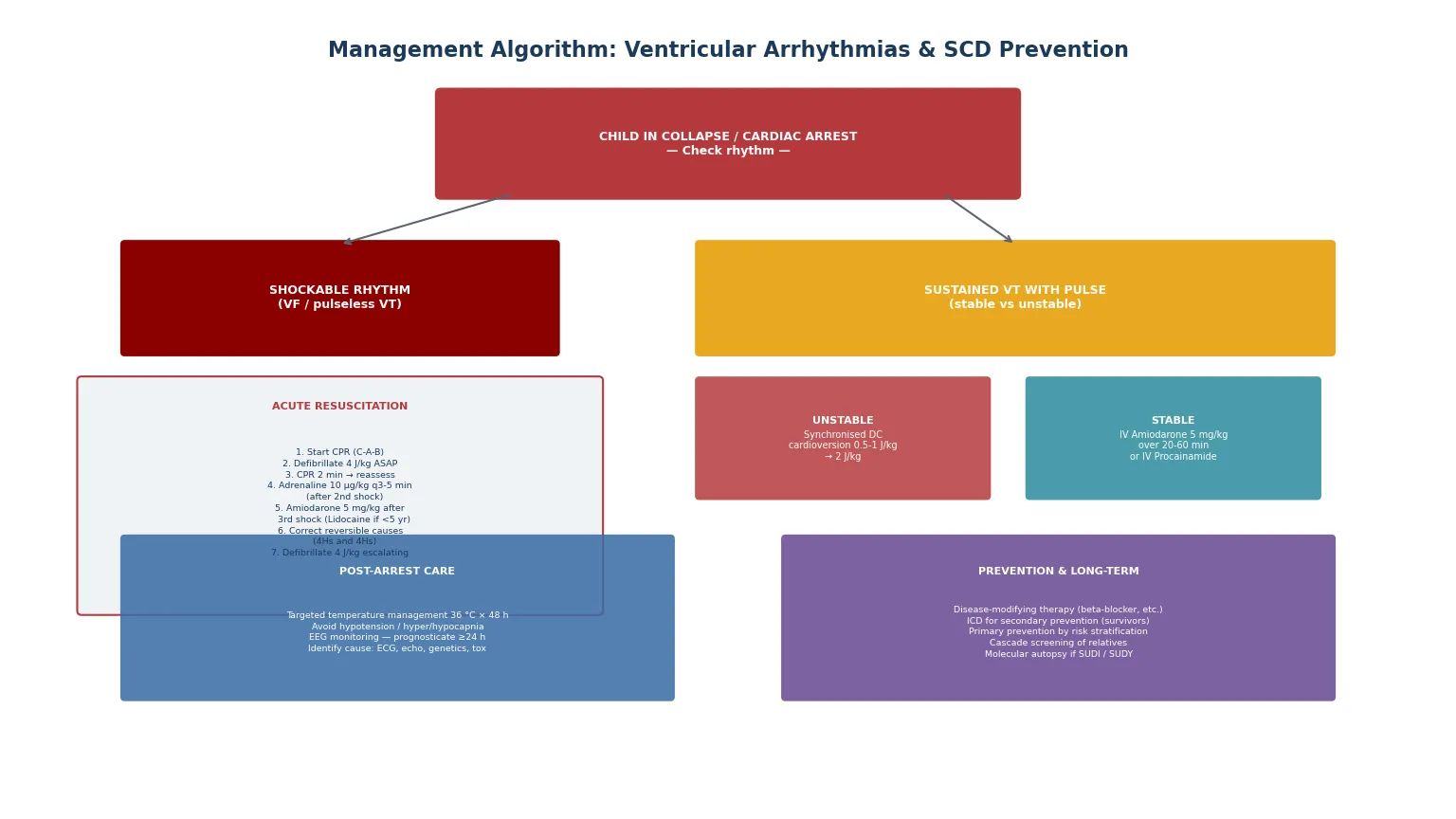
For a child in ventricular fibrillation or pulseless ventricular tachycardia, begin cardiopulmonary resuscitation immediately (chest compressions at one hundred to one hundred and twenty per minute, compression-to-ventilation ratio fifteen-to-two for two rescuers, thirty-to-two for single rescuers) and defibrillate as soon as possible at two to four joules per kilogram, escalating up to a maximum of ten joules per kilogram or the adult dose if required. After each shock, resume CPR immediately for two minutes before reassessing the rhythm — pausing compressions to check the rhythm is a common and damaging error. Give adrenaline at ten micrograms per kilogram every three to five minutes (after the second shock), and give amiodarone at five milligrams per kilogram after the third shock if VF or pVT persists. Lidocaine at one milligram per kilogram is an alternative antiarrhythmic, and is preferred over amiodarone in children under five years by some guidelines. Throughout, identify and correct the reversible causes (the four Hs and four Ts: hypoxia, hypovolaemia, hypo- or hyperkalaemia, hypothermia; thrombosis, tamponade, tension pneumothorax, toxins). [3] [4]
SHOCKABLE
For a child with sustained VT and a pulse, the management divides at haemodynamic stability. An unstable child — shock, hypotension, altered consciousness, severe heart failure — requires synchronised DC cardioversion at half to one joule per kilogram, escalating to two joules per kilogram if the first shock fails, with sedation if time permits but without delay if the child is compromised. A stable child — preserved perfusion, normal mental state — can be treated pharmacologically with intravenous amiodarone at five milligrams per kilogram over twenty to sixty minutes, or intravenous procainamide as an alternative. Correct electrolyte abnormalities, stop any causative drugs, and arrange urgent transfer to a cardiac centre for definitive diagnosis and management. [2] [3]
Management — Definitive & Stepwise
Definitive management is stratified by the underlying cause and by the risk of recurrent arrhythmia, and the modern approach has four pillars: disease-modifying therapy (the beta-blocker for the channelopathy, the aldosterone antagonist or heart failure therapy for the cardiomyopathy, the surgical or catheter intervention for the congenital lesion), catheter ablation for the focal or isthmus-dependent VT, the implantable cardioverter defibrillator for the high-risk patient, and the lifestyle and surveillance package. The goal is to reduce arrhythmic events while minimising the burden of therapy, which is particularly important in children. [3] [4]
The life arc of a child after a ventricular arrhythmia event
Post-arrest care is now an integral part of the resuscitation, not an afterthought. Targeted temperature management — maintaining the core temperature at thirty-six degrees Celsius for forty-eight hours after return of spontaneous circulation — is the recommended post-arrest strategy, with avoidance of fever. Haemodynamic optimisation is critical: post-resuscitation hypotension is strongly associated with mortality, and the mean arterial pressure should be maintained at age-appropriate targets with fluid, inotropes, or vasopressors as needed. Avoid hyperoxia (wean the fractional inspired oxygen to the lowest level maintaining adequate saturation) and avoid both hypercapnia and hypocapnia (target normocapnia). Continuous EEG monitoring is essential because subclinical seizures are common after cardiac arrest, and prognostication should not be attempted before twenty-four hours of normothermia. [7]
The implantable cardioverter defibrillator is the definitive therapy for the patient at high risk of recurrent life-threatening ventricular arrhythmia. Secondary prevention ICDs are indicated for survivors of cardiac arrest due to VF or haemodynamically unstable VT in whom a reversible cause has been excluded — these patients have a high rate of recurrence. Primary prevention ICDs are considered for patients with risk factors for sudden death (hypertrophic cardiomyopathy with specific high-risk features, arrhythmogenic right ventricular cardiomyopathy with sustained VT, some forms of dilated cardiomyopathy with reduced ejection fraction, and channelopathy patients with high-risk features). In children, the threshold for ICD implantation is higher than in adults because of the technical challenges (lead fractures from growth and activity, infection, the need for generator and lead revisions over a lifetime) and the psychological burden of inappropriate shocks, which are more common in young patients whose physiological heart rates may overlap with detection zones. Subcutaneous and extravascular ICD systems are expanding the options for children. [3] [4]
Catheter ablation is the definitive therapy for idiopathic VT with a focal origin (right ventricular outflow tract VT, fascicular VT) or an anatomical isthmus (post-surgical tetralogy of Fallot VT), and it can be curative, avoiding the need for lifelong antiarrhythmic drugs or an ICD. The success rate depends on the substrate: focal idiopathic VT has a high ablation success rate, while post-surgical re-entry VT may require extensive substrate mapping and has a higher recurrence rate. [2] [4]
Specific Subtypes & Scenarios
Post-surgical tetralogy of Fallot VT is the classic late arrhythmic complication of congenital heart disease repair, presenting years to decades after the operation with sustained monomorphic VT from re-entry around the right ventricular outflow tract patch, the ventriculotomy scar, or the conal septum. Risk factors include older age at repair, a transannular patch, significant pulmonary regurgitation, right ventricular dilatation, and QRS duration above one hundred and eighty milliseconds on the ECG. Management includes surgical pulmonary valve replacement (which reduces the right ventricular volume load but does not reliably eliminate the arrhythmia substrate), catheter ablation of the critical isthmus, and ICD implantation for the highest-risk patients. [2] [4]
Idiopathic ventricular fibrillation describes cardiac arrest due to VF in a patient with no identifiable structural, channelopathic, metabolic, or toxic cause after comprehensive evaluation. It accounts for a proportion of sudden arrhythmic death syndrome cases, and some are subsequently found to have rare genetic causes (e.g., calmodulin mutations, DPP6 variants) on extended genetic testing. Management is an ICD for secondary prevention, because the recurrence rate is significant. The early repolarisation pattern on the ECG has been associated with idiopathic VF in some cohorts, but its role in risk stratification in children is uncertain. [10]
Commotio cordis is the instantaneous production of ventricular fibrillation by a blunt, non-penetrating chest-wall impact, with no underlying structural cardiac injury. It is most common in adolescent males in projectile sports (baseball, lacrosse, hockey, martial arts) and the critical determinants are the timing of the impact on the cardiac cycle (the vulnerable repolarisation window), the force, and the properties of the projectile. Survival has improved dramatically with immediate CPR and AED access at sporting venues, and prevention includes the use of softer safety baseballs, chest protectors (though their efficacy is debated), and AED availability at all organised sporting events. [5] [6]
Drug-induced ventricular arrhythmia is most commonly torsades de pointes from QT-prolonging medications (macrolide and fluoroquinolone antibiotics, ondansetron, antipsychotics, methadone) in a child with either congenital long-QT syndrome or acquired QT prolongation from electrolyte disturbance or drug interaction. Sodium-channel-blocker toxicity (tricyclic antidepressants, class I antiarrhythmics) produces a different picture of wide-QRS tachycardia and sodium-channel-blocker toxicity, managed with intravenous sodium bicarbonate. Every child resuscitated from an arrhythmic event must have a thorough medication and toxicology review. [2] [12]
Cardiac arrest from myocarditis may be the first presentation of fulminant myocarditis in a previously well child, with ventricular arrhythmia and cardiogenic shock. The management combines the standard resuscitation with mechanical circulatory support (extracorporeal membrane oxygenation) for the refractory case, because some children with fulminant myocarditis recover fully if supported through the acute phase. Eosinophilic or giant-cell myocarditis has a particularly poor prognosis and may require urgent transplantation. [2] [4]
Complications & Pitfalls
The complications of ventricular arrhythmia are the arrhythmic events themselves — syncope, aborted cardiac arrest, and sudden cardiac death — and the complications of therapy. On the disease side, the untreated patient at risk carries an annual event rate that varies enormously with the underlying condition: secondary prevention patients (survivors of cardiac arrest) have a recurrence rate of five to fifteen per cent per year, while the primary prevention patient's risk depends on the substrate and the risk factors identified. [3] [4]
On the therapy side, the ICD brings a distinct set of complications that are more burdensome in children than in adults: lead fractures (from growth and activity, requiring lead revision), infection, inappropriate shocks (the paediatric heart rate may overlap with detection zones during exercise or sinus tachycardia, and atrial arrhythmias may be misdetected as ventricular), and the psychological impact of living with a device (anxiety, depression, restrictions, and the impact on body image and relationships). Amiodarone, used chronically in some patients, brings thyroid dysfunction, pulmonary fibrosis, photosensitivity, corneal microdeposits, and hepatotoxicity, and is used sparingly in children for this reason. [3]
The risks that drive management intensity
The common clinical pitfalls are: treating a wide-QRS tachycardia as supraventricular tachycardia with aberrancy in a haemodynamically compromised child (the safe default is VT until proven otherwise); delaying defibrillation in VF or pulseless VT while waiting for drug access or specialist arrival (defibrillate early); pausing CPR to check the rhythm after a shock (resume compressions immediately); failing to obtain a rhythm strip or ECG in a child presenting with collapse; misdiagnosing arrhythmic syncope as epilepsy or vasovagal; failing to identify a reversible cause (electrolytes, drugs, ischaemia) in the resuscitation; and failing to arrange cascade screening of relatives after a diagnosis or a sudden death. The psychosocial burden of an arrhythmia or ICD diagnosis — anxiety, restrictions, body image, the impact on siblings and parents — is also a pitfall if unaddressed. [1] [8]
Prognosis & Disposition
The prognosis after a ventricular arrhythmia event depends on the underlying cause, the quality of the resuscitation, and the effectiveness of the long-term therapy. Survival to discharge after out-of-hospital cardiac arrest from VF or pulseless VT in children is approximately twenty-five to forty per cent, and it is strongly determined by the time to first shock, the quality of bystander CPR, and the quality of post-arrest care. Neurological outcome is the other critical determinant, and it is improved by high-quality CPR, early defibrillation, targeted temperature management, and haemodynamic optimisation. [3] [7]
With modern ICD therapy, the long-term prognosis for the survivor of cardiac arrest is favourable in terms of arrhythmic death, because the device effectively terminates recurrent VF or VT. The challenges are the complications of the device, the psychosocial burden, and the management of the underlying condition. For the patient with a preventable substrate — the channelopathy on effective beta-blockade, the cardiomyopathy on disease-modifying therapy, the post-surgical congenital heart disease after pulmonary valve replacement and ablation — the outlook is good with appropriate surveillance and therapy. [3] [4]
Disposition after an acute event is PICU or high-dependency care for the immediate post-arrest period, transfer to a specialist paediatric cardiac centre for the diagnostic workup and the risk-stratification decision, and outpatient management under a specialist inherited cardiac conditions and electrophysiology service for the long term. The general paediatrician plays a critical role in co-ordination, adherence support, the management of intercurrent illness (which can destabilise the arrhythmia substrate through fever, electrolyte disturbance, and drug interactions), and the psychosocial support of the child and family. [2] [4]
Special Populations
Neonates and infants with ventricular arrhythmia most commonly have benign accelerated idioventricular rhythm or isolated ventricular ectopy, both of which resolve spontaneously and require only documentation and reassurance if the heart is structurally normal and the ectopy is monomorphic and low-burden. Severe early-onset VT or VF is rare but may be due to calmodulin mutations (causing recurrent cardiac arrest in infants with extreme QT prolongation), congenital long-QT syndrome presenting with two-to-one atrioventricular block, or an inborn error of metabolism. [2] [12]
Children with congenital heart disease — particularly those who have undergone repair of tetralogy of Fallot, transposition of the great arteries, or aortic stenosis — require lifelong cardiac surveillance with ECG, ambulatory monitoring, and exercise testing to detect the arrhythmia substrate before it produces an event. The post-ToF patient is the archetype, and the QRS duration on the ECG is a useful surrogate marker for the risk of sustained VT (a QRS above one hundred and eighty milliseconds carries a significantly increased risk). [2] [4]
The transitioning adolescent and young adult faces the convergence of several risks: adherence challenges with medication and follow-up, the highest-risk period for events (adolescence and the early twenties), recreational drug and alcohol use (cocaine and stimulants can trigger ventricular arrhythmia), and the transition to adult care. Structured transition programmes — education, self-management, and a warm handover to the adult inherited cardiac conditions and electrophysiology service — are now standard. Pregnancy in the patient with an ICD or channelopathy requires planning, because the haemodynamic changes of pregnancy can destabilise the arrhythmia, and the ICD may need reprogramming for the higher baseline heart rate. [3] [4]
Resource-limited and remote populations — including Indigenous, refugee, and rural communities — face diagnostic delays and limited access to specialist services, genetic testing, and ICD therapy. Telehealth cardiology, protocol-driven beta-blocker management, a low threshold for referral after any exertional syncope, and investment in bystander CPR and AED access in schools and public spaces are the pillars of equitable practice. The global reality is that most children at risk of SCD are never identified before the index event, and the index event is too often fatal. [1] [2]
Evidence, Guidelines & Regional Differences
| Region | Key guideline | Acute antiarrhythmic | Screening approach |
|---|
The controversies a candidate should be able to discuss are the optimal antiarrhythmic in paediatric VF (amiodarone versus lidocaine, with some evidence favouring lidocaine in younger children), the role of routine preparticipation ECG screening for athletes (favoured in Europe, debated in North America and ANZ), the threshold for primary prevention ICD implantation in children with cardiomyopathy or channelopathy (higher than in adults, individualised), the approach to the genotype-positive asymptomatic child (individualised by condition), and the role of the subcutaneous and extravascular ICD in children (expanding, particularly for the primary prevention patient in whom transvenous leads are problematic). [3] [4]
Exam Pearls
References
- [1]Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med, 2016.PMID 27332903
- [2]Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace, 2015.PMID 26318695
- [3]Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. J Am Coll Cardiol, 2018.PMID 29097296
- [4]Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J, 2022.PMID 36017572
- [5]Maron BJ, Haas TS, Ahluwalia A, Garberich RF, Estes NA 3rd, Link MS Commotio cordis and the epidemiology of sudden death in competitive lacrosse. Pediatrics, 2009.PMID 19706581
- [6]Madias C, Maron BJ, Weinstock J, Estes NA 3rd, Link MS Commotio cordis. Indian Pacing Electrophysiol J, 2007.PMID 17957272
- [7]Topjian AA, Telford R, Birnkrant DJ, Burns JP, Gilbert K, et al. Association of early postresuscitation hypotension with survival to discharge after pediatric cardiac arrest. JAMA Pediatr, 2018.PMID 29228147
- [8]Abrams DJ, Macrae CA Long QT syndrome. Circulation, 2014.PMID 24709866
- [9]Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation, 2000.PMID 10673253
- [10]Conte G, Sieira J, Ciconte G, de Asmundis C, Chierchia GB, et al. Out-of-hospital cardiac arrest due to idiopathic ventricular fibrillation in patients with normal electrocardiograms: results from a multicentre long-term registry. Europace, 2019.PMID 31504477
- [11]Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation, 2004.PMID 15051644
- [12]Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm, 2013.PMID 24011539