Paeds · clinical-pharmacology-and-therapeutics
Endocrine and diabetes medicines
Also known as Paediatric endocrine prescribing · Insulin therapy in children · Levothyroxine in children · Recombinant growth hormone therapy · Diabetes and thyroid medicines in childhood
A fellowship approach to the three highest-yield paediatric endocrine medicine families: insulin for type 1 diabetes and diabetic ketoacidosis, levothyroxine for congenital and acquired hypothyroidism, and recombinant human growth hormone for growth hormone deficiency and licensed growth-failure indications. Covers insulin pharmacokinetic classes, the 0.05 to 0.1 unit per kg per hour DKA infusion, basal-bolus and pump regimens, ISPAD 2024 glycemic targets, neonatal levothyroxine 10 to 15 microgram per kg per day, and recombinant growth hormone 0.045 to 0.050 mg per kg per day with monitoring and safety surveillance from neonatal life through transition.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child walks in. The endocrine axis that quietly ran their metabolism has failed, and the fix is to give back the missing hormone from the outside. Endocrine prescribing is the deliberate act of choosing the right hormone preparation, the right weight-based dose, the right timing, and the right monitoring — so that the child grows, metabolises and develops as if the gland were still working. [1]
Three medicine families carry almost all of the exam weight, and they are the ones a general paediatrician writes in everyday practice rather than leaving entirely to the specialist. Insulin replaces the hormone the beta-cells can no longer make in type 1 diabetes and is the definitive, switch-off-ketogenesis drug in diabetic ketoacidosis. Levothyroxine replaces the thyroxine the thyroid cannot make in congenital and acquired hypothyroidism, and in the first two years of life it is a neurodevelopmental drug as much as a metabolic one. Recombinant human growth hormone drives linear growth in growth hormone deficiency and a handful of licensed growth-failure indications, and it is a daily, self-injected, years-long commitment that tests adherence and safety surveillance at every visit. [1] [6]
The reason these three dominate is that they are common, they are dangerous when misprescribed, and they sit at the centre of the fellowship written and clinical exams. A child in DKA given an insulin bolus before fluids can die of cerebral oedema; a neonate whose levothyroxine is delayed past the first weeks loses IQ points that never come back; an adolescent on growth hormone with a new headache may have intracranial hypertension. The fellowship candidate is expected to write the dose, defend the regimen, and explain the safety-net — not to defer the whole problem to an endocrinologist. [4] [6]
Classification
You cannot prescribe endocrine medicines sensibly until you classify them by pharmacokinetic behaviour and licensed indication. Mix the classes and you will write a regimen that looks defensible and behaves unpredictably. [1]
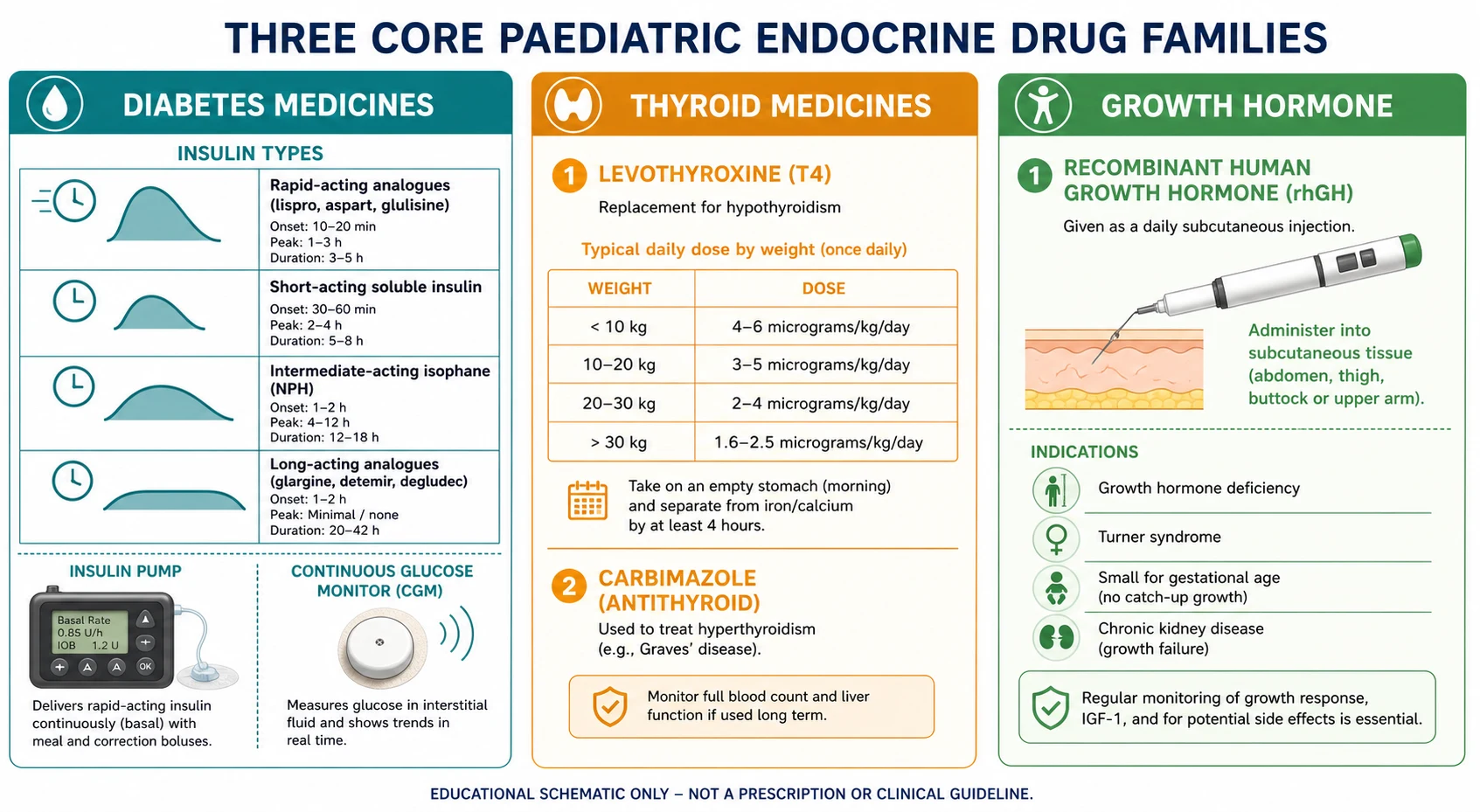
Insulin is classified by how fast it acts and how long it lasts. Rapid-acting analogues (lispro, aspart, glulisine) start within 10 to 20 minutes, peak at 1 to 3 hours and last 3 to 5 hours — these are meal-time or bolus insulins and the only insulins used in a pump. Short-acting soluble human insulin starts in 30 minutes, peaks at 2 to 5 hours and lasts up to 8 hours; it is the form used intravenously in DKA. Intermediate-acting isophane (NPH) peaks at 4 to 12 hours and lasts 12 to 18 hours; it was the original basal insulin and is still used where analogues are not funded. Long-acting analogues (glargine, detemir, degludec) have a flat, peakless profile lasting around 24 hours (longer for degludec) and are the modern basal insulins. The pairing that defines modern therapy is one daily basal plus a rapid-acting bolus with each meal — the basal-bolus regimen. [1]
Thyroid medicines are simpler. Levothyroxine (T4) is the standard replacement: synthetic, orally absorbed, converted peripherally to the active T3, and titrated to a suppressed TSH in primary hypothyroidism. Liothyronine (T3) is reserved for rare situations and is not first-line in children. Carbimazole sits on the opposite side of the axis as the antithyroid drug for hyperthyroidism, with propylthiouracil and block-and-replace or titration regimens as the specialist choices. [6]
Recombinant human growth hormone (somatropin) is a single drug with several licensed indications, and the licence is what the examiner wants you to name. The funded indications include growth hormone deficiency, Turner syndrome, Prader-Willi syndrome, small for gestational age children who have not caught up by age 4, chronic kidney disease with growth failure, SHOX deficiency, and in some jurisdictions idiopathic short stature. The dose is weight-based, and the indication changes the monitoring and the counselling but not the injection. [8] [12]
Epidemiology & Risk Factors
Endocrine medicines reach a remarkably wide cross-section of children, which is why a general paediatrician cannot delegate them entirely. Type 1 diabetes is the commonest chronic endocrine disease of childhood, and its incidence is rising in the under-fives — the very children in whom an insulin dose is hardest to write because eating is unpredictable. Diabetic ketoacidosis remains a leading cause of diabetes-related death in children, and most of those deaths are tied to cerebral oedema and to prescribing decisions about fluids and insulin. [4] [9]
Congenital hypothyroidism affects roughly 1 in 2000 to 1 in 4000 newborns, and because newborn screening catches it before symptoms, levothyroxine becomes a time-critical prescription in an asymptomatic neonate. The neurodevelopmental window is narrow: thyroid hormone is essential for brain myelination in the first two years, and a delayed or under-dosed replacement costs IQ points that do not return. [6] [7]
Growth hormone deficiency and the licensed growth-failure indications together account for a smaller but highly monitored group. Recombinant growth hormone is a daily subcutaneous injection, given for years, at high cost, with a heavy adherence burden — which is why prescribing it is as much a family-capacity and counselling decision as a hormonal one. [8] [12]
The highest prescribing risk concentrates in recognisable groups: very young children with erratic eating and activity; adolescents with variable adherence, disordered eating, and puberty-related insulin resistance; children in DKA at presentation; and children with coexisting autoimmunity such as coeliac disease or thyroiditis, which themselves change insulin requirement and growth. [1] [9]
Pathophysiology
To prescribe endocrine medicines with understanding rather than by rote, you need the feedback loops the drugs are built to restore. Each of the three families replaces a hormone at a different point in a different loop, and the dose you write is really a calculation of how much external hormone is needed to put the loop back in balance. [6]
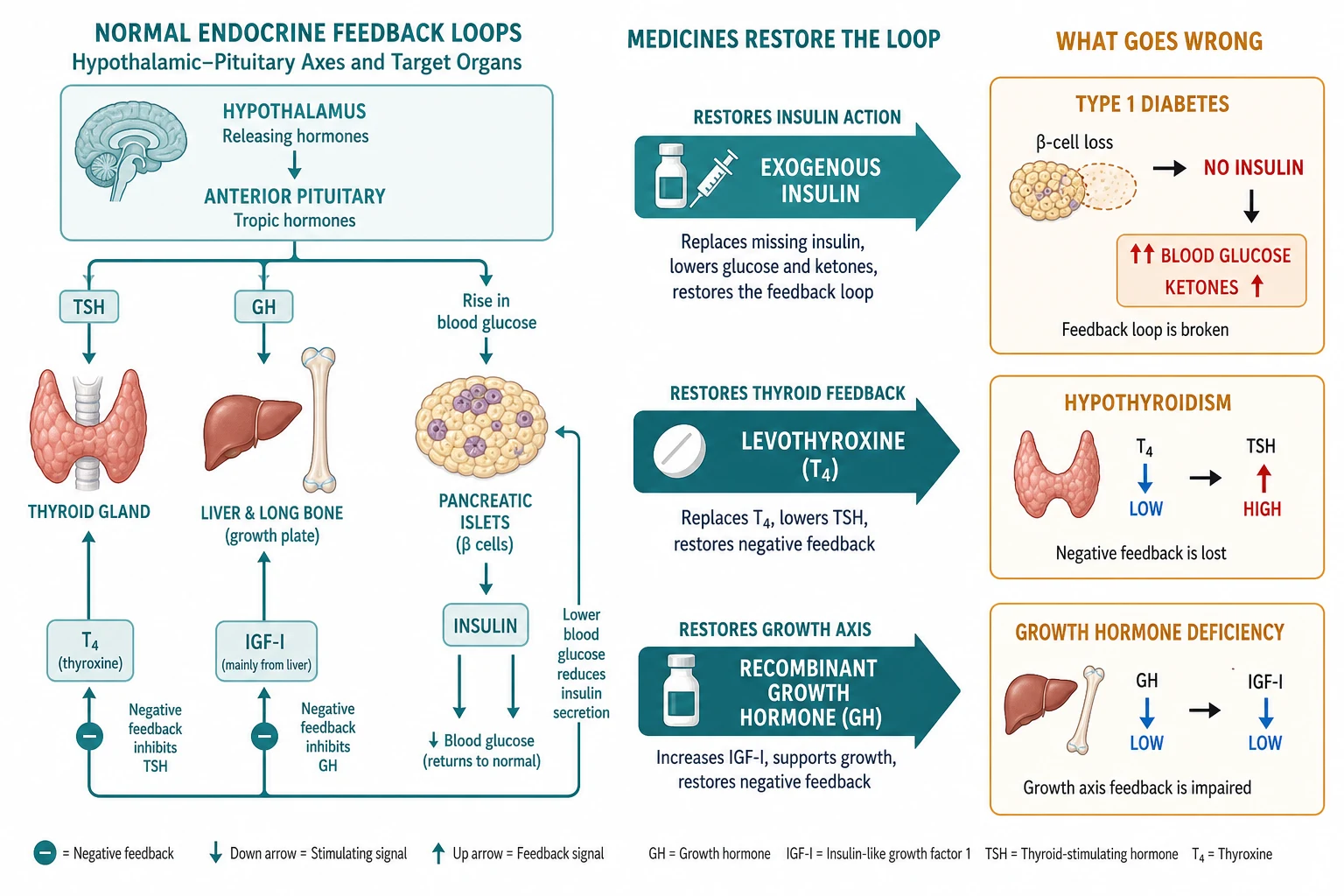
Insulin and the pancreatic islet. Type 1 diabetes is autoimmune destruction of the beta-cells, producing absolute insulin deficiency. Without insulin, glucose cannot enter muscle and fat, so glucose rises; at the same time lipolysis runs unchecked, flooding the liver with free fatty acids that it converts to ketone bodies. The blood becomes acidic. Exogenous insulin reverses both arms at once: it restores glucose uptake, suppressing hyperglycaemia, and it switches off lipolysis, stopping ketogenesis. That dual action is why insulin — not fluids, not bicarbonate — is the definitive treatment of DKA. [1] [4]
Cerebral oedema is the feared complication of paediatric DKA, and the mechanism is still debated but probably combines cerebral hypoperfusion during dehydration, reperfusion on treatment, and the osmotic shift as glucose falls. The practical consequence is the prescribing rule that shapes the whole protocol: resuscitate the volume first with isotonic saline, then start insulin, avoid boluses, and keep the glucose fall gradual by adding dextrose once the level falls. [9]
Levothyroxine and the thyroid axis. The hypothalamus releases TRH, which drives the pituitary to release TSH, which drives the thyroid to make T4 and a little T3. T4 feeds back negatively on the pituitary. In primary hypothyroidism the thyroid fails, T4 falls, TSH rises, and the child slows metabolically. Levothyroxine is synthetic T4: you give enough to normalise the TSH, and the negative-feedback loop does the rest. In the first two years, thyroid hormone is also a direct neurodevelopmental signal, which is why the dose and the timing matter so much in infancy. [6]
Growth hormone and the growth axis. Hypothalamic GHRH drives the pituitary to secrete growth hormone in pulses, mostly overnight. Growth hormone acts on the hepatic and tissue growth-hormone receptor to produce IGF-I, and IGF-I drives linear growth at the growth plate while feeding back on the pituitary. Recombinant growth hormone is given as a daily subcutaneous injection to reproduce the physiological pulse, and the response you titrate to — growth velocity and IGF-I — is the downstream readout of that same axis. [12]
Clinical Presentation
The child harmed by a misprescribed endocrine medicine rarely arrives with the drug written on their forehead. They arrive with deterioration, and the medicine is found when someone thinks to look. [4]
A child with new or poorly controlled type 1 diabetes classically has the four Ts of polyuria, polydipsia, weight loss and tiredness — often preceded by nocturia or new enuresis in a previously dry child, and sometimes by recurrent thrush. The trap is the toddler who presents for the first time already in DKA: vomiting, abdominal pain, deep sighing Kussmaul breathing, dehydration and a falling conscious level. The first job is to recognise DKA; the prescribing decisions then follow. [4] [9]
Hypoglycaemia in a child on insulin is the bedside emergency the family must be able to recognise and treat. It presents with sweating, tremor, hunger, confusion, and at the extreme seizure or collapse. The prescribing lesson is that a severe hypo always changes the next insulin dose and the sick-day plan, and that the family needs a glucagon supply and a clear rule for fast-acting carbohydrate. [1]
A neonate with congenital hypothyroidism is usually asymptomatic at birth because maternal thyroid hormone crosses the placenta; the diagnosis comes from the newborn screen, and the medicine must follow the screen rather than the symptoms. If treatment is delayed, the infant then develops the classical signs — prolonged jaundice, constipation, a large fontanelle, hypotonia, a hoarse cry and poor feeding — and the neurodevelopmental cost is already accruing. [6]
A child on levothyroxine who is over-replaced is agitated, heat-intolerant, tachycardic and growing poorly or losing weight; a child who is under-replaced is lethargic, constipated, cold, slow and growing poorly. Growth is the common signal that the dose is wrong, which is why weight and height are checked at every thyroid visit. [6]
A child on growth hormone who responds well shows a catch-up in growth velocity in the first year. The prescribing red flag is the safety trio: a new headache, vomiting or visual change raises intracranial hypertension; a limp raises slipped capital femoral epiphysis; and unexpected hyperglycaemia raises glucose intolerance. Each demands a held dose and an assessment, not a phone-adjusted increase. [12]
Differential Diagnosis
When an endocrine medicine is not working, the first question is not "what is wrong with the drug" but "is this the right drug, the right dose, or the wrong diagnosis." Three distinctions carry most of the weight. [1]
First, in a child on insulin with abnormal glucose or HbA1c, distinguish a regimen problem from adherence, intercurrent illness, puberty and coexisting disease. A regimen problem means the insulin type, dose or technique is wrong — injection into lipohypertrophic tissue, an expired pen, a basal given at the wrong time, or an insulin-to-carbohydrate ratio that no longer fits. Coeliac disease and thyroiditis both raise insulin requirement and must be screened. [1]
Second, distinguish type 1 diabetes from type 2, monogenic diabetes (MODY) and secondary diabetes, because the medicine changes. The child with type 2 may need metformin and weight management rather than basal-bolus insulin; the child with neonatal monogenic diabetes may respond to sulfonylureas and need no insulin at all. Antibody testing and a C-peptide settle the question when the phenotype is unclear. [1]
Third, before committing a child to a lifelong endocrine drug, confirm the diagnosis is what you think. Congenital hypothyroidism must be separated from transient hypothyroidism, hyperthyrotropinaemia, iodine deficiency, central hypothyroidism and the sick-euthyroid state. Growth failure must be separated from familial short stature, constitutional delay of growth and puberty, primordial short stature, hypothyroidism, coeliac disease, Turner syndrome and chronic disease before growth hormone is started. [6] [8]
Separate DKA from its mimics — sepsis, gastroenteritis, pneumonia, metabolic acidosis and salicylate toxicity — because the child in ketosis with acidosis and hyperglycaemia needs insulin first, and the child with sepsis and a normal glucose needs antibiotics and fluids. A bedside glucose and ketone reading resolves most of the ambiguity in minutes. [4]
Clinical & Bedside Assessment
The endocrine prescribing assessment is a bedside act, not a laboratory one. The numbers matter, but they only matter once you have examined the child, read the chart, and watched how the family uses the device. [1]
At the bedside of a child on insulin, weigh the child, look at the injection sites for lipohypertrophy (the commonest reversible cause of erratic control), assess hydration and conscious level, and ask about intercurrent illness, technology use and the glucose pattern over the last two weeks. In a suspected DKA, grade the dehydration, check the GCS, look for shock and Kussmaul breathing, and take a bedside glucose and ketone. The prescribing decision — fluids first, then insulin — flows directly from this assessment. [4]
At the bedside of a child on levothyroxine, weigh and measure the child, plot growth, stage puberty, and examine the heart for the tachycardia of over-replacement. Ask specifically about adherence, because missed doses are the commonest reason for a rising TSH, and ask whether the dose is given on an empty stomach, since food and calcium or iron supplements reduce absorption. [6]
At the bedside of a child on growth hormone, measure height and calculate growth velocity, stage puberty, and ask directly about headache, visual change and limp. Check the injection technique with the family, because a growth response that flattens often traces to technique or missed doses rather than to dose inadequacy. Look at the IGF-I trend, which tells you whether you are under- or over-shooting. [12]
How you communicate the dose change matters as much as the change itself. The carer must be able to repeat the dose, draw up or dial the device, and explain the sick-day and hypoglycaemia plans. The adolescent needs private time to discuss adherence, driving, alcohol and sexual health without the parent in the room — a structured part of every transition visit. [1]
Investigations
The investigations for endocrine prescribing are the investigations of control: glucose and HbA1c for insulin, TSH and free T4 for levothyroxine, and growth velocity with IGF-I for growth hormone. Each has a target, a timing, and an action that flows from the result. [1]
For insulin prescribing, capillary glucose is the day-to-day tool, HbA1c is the 3-monthly summary, and continuous glucose monitor (CGM) metrics — time in range and the glucose management indicator — increasingly complement both. The ISPAD 2024 glycemic targets set an HbA1c below 7 percent for most children, individualised to age and hypoglycaemia risk, with time-in-range goals used alongside capillary readings. Ketones (blood beta-hydroxybutyrate preferred over urine) are the DKA and sick-day marker that drives the insulin decision. [2] [3]
For levothyroxine, TSH is the primary target in primary hypothyroidism, with free T4 used when central hypothyroidism is suspected or in infancy where a suppressed TSH can lag. Recheck at 2 and 4 weeks after a dose change, then every 1 to 3 months in the first two years and every 3 to 6 months thereafter. A TSH in the age-appropriate reference range confirms adequate replacement. [6]
For growth hormone, the response is read from growth velocity (the centile crossing of a well-measured height over 6 to 12 months) and IGF-I (the biochemical marker that guides dose titration into the reference range). Bone age, thyroid function and a diagnostic stimulation test inform the decision to start, while the minimum monitoring dataset — growth, IGF-I, glucose, thyroid function and safety review — frames every follow-up visit. [8] [12]
Management — Resuscitation
When a child on an endocrine medicine presents in crisis, the crisis is managed first and the dose arithmetic second. The two resuscitation scenarios a general paediatrician must run without hesitation are diabetic ketoacidosis and severe hypoglycaemia. [4]
In DKA the sequence is airway, breathing and circulation, then fluid resuscitation with isotonic saline before any insulin, then intravenous insulin. Give a 10 to 20 mL per kg isotonic saline bolus only if the child is in shock, otherwise begin a calculated deficit replacement over 24 to 48 hours. Once intravascular volume is restored, start a continuous intravenous insulin infusion at 0.05 to 0.1 unit per kg per hour using soluble or rapid-acting insulin. Never give an intravenous insulin bolus in paediatric DKA, because a bolus drives a steep glucose and osmolar fall that contributes to cerebral oedema. Add dextrose to the fluids when the glucose falls to around 11 to 14 mmol per L, keep the potassium replaced, and transition back to subcutaneous insulin only when the ketones have cleared and the child is eating. [4] [9]
Severe hypoglycaemia in a child on insulin is treated by stopping insulin, giving 15 g of fast-acting carbohydrate if the child is conscious, or intravenous dextrose 0.1 to 0.3 g per kg (2 to 5 mL per kg of 10 percent dextrose) or glucagon 0.5 mg intramuscular (1 mg for the older child) if the child is unconscious. Every severe hypo then triggers a review of the insulin regimen, the sick-day plan, and the family's ability to manage at home. [1]
A neonate with a confirmed congenital hypothyroidism screen is treated immediately — levothyroxine at 10 to 15 microgram per kg per day, started within the first two weeks of life, because every week of thyroid hormone deficit in infancy erodes neurodevelopment. This is a resuscitation-grade time pressure even though the child looks well. [6] [7]
A child on growth hormone who develops severe headache, vomiting or visual change is assessed for intracranial hypertension: hold the growth hormone, examine the fundi, and arrange imaging. The dose is not increased while the symptom is unexplained. [12]
Management — Definitive & Stepwise
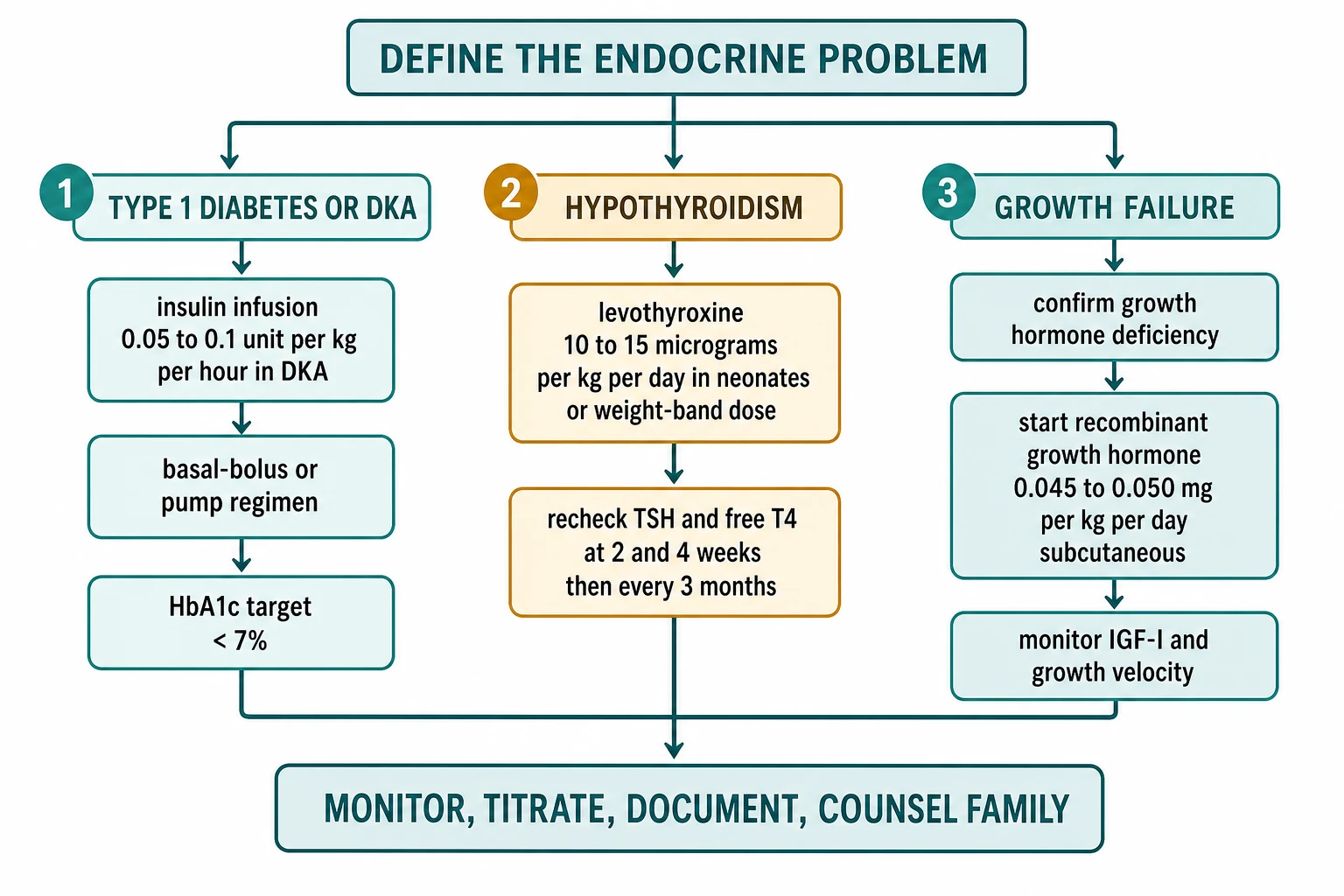
Once the child is stable, endocrine prescribing follows a reproducible algorithm: name the axis that is failing, choose the hormone preparation, set the weight-based dose, plan the monitoring, and close the loop with documentation and counselling. Run it the same way every time and you will not miss a step. [1]
Insulin for type 1 diabetes
For a child with newly diagnosed type 1 diabetes who is not in DKA, set the total daily dose at about 0.5 to 0.75 unit per kg per day, often lower in the partial-remission honeymoon phase. Split it roughly half basal and half bolus. A typical basal-bolus regimen is a once-daily long-acting analogue (glargine, detemir or degludec) plus a rapid-acting analogue (lispro, aspart or glulisine) with each meal, using an insulin-to-carbohydrate ratio for food and a correction factor for high glucose. Titrate the basal for fasting trends and the ratios for post-meal excursions, reading the pattern from the CGM or the glucose diary over several days rather than reacting to a single number. [1] [2]
When a pump or hybrid closed-loop system fits the child and the family, it can improve time in range and reduce hypoglycaemia, and the evidence in children and adolescents is now strong. The choice depends on age, adherence, hypoglycaemia burden, access, funding and family capacity — not on a single best device. Whatever the platform, the family needs a pump-failure and ketone plan, because an interrupted pump causes DKA within hours when there is no subcutaneous basal reserve. [5] [11]
Levothyroxine for hypothyroidism
For a neonate with congenital hypothyroidism, start at 10 to 15 microgram per kg per day once daily, ideally on an empty stomach, and recheck TSH and free T4 at 2 and 4 weeks then every 1 to 3 months in infancy. For older children, use weight-band dosing — roughly 5 to 6 microgram per kg per day in a 1 to 5 year old and 4 to 5 microgram per kg per day in a 6 to 12 year old — trending toward the adult dose of about 1.6 microgram per kg per day in late adolescence. Recheck after each change and at least every 3 to 6 months through growth, because the dose must grow with the child. [6] [7]
Recombinant growth hormone for growth failure
For a child with confirmed growth hormone deficiency or a licensed indication, start at 0.045 to 0.050 mg per kg per day (about 0.15 to 0.17 IU per kg per week) subcutaneous once daily, usually at bedtime to mirror the physiological overnight pulse. Titrate to an IGF-I in the age-appropriate reference range and to the growth response, reviewing every 3 to 6 months. Run the safety surveillance at each visit — ask about headache and visual change (intracranial hypertension), examine for limp (slipped capital femoral epiphysis), and check glucose and thyroid function. Continue to near-adult height, then reassess for adult growth hormone deficiency at transition. [8] [12]
Name the failing axis: pancreatic islet, thyroid, or growth hormone axis
Choose the hormone preparation (insulin class, levothyroxine, recombinant GH)
Set the weight-based dose from the current guideline or formulary (ISPAD, ESPE, BNFc, NICE, RCH Melbourne)
Plan the monitoring: glucose or HbA1c; TSH and free T4; growth velocity and IGF-I
Close the loop: document the regimen, teach the technique, supply a sick-day and hypoglycaemia plan, counsel the family, arrange specialist follow-up
Specific Subtypes & Scenarios
The algorithm is constant; the scenarios stress different parts of it. Walk through the high-yield ones the way an examiner will. [1]
Neonate with congenital hypothyroidism on newborn screening. Confirm the diagnosis, start levothyroxine at 10 to 15 microgram per kg per day within two weeks, and recheck TSH and free T4 at 2 and 4 weeks. Counsel the family that the first two years are the neurodevelopmental window, that the dose will rise as the baby grows, and that transient disease is re-tested later, not assumed now. [6] [7]
Toddler with new type 1 diabetes. Unpredictable eating and activity make insulin dosing hard, so prefer a regimen that is forgiving, teach injection-site rotation from day one, individualise the HbA1c target, and build family capacity with structured education. The very young child is also the one at highest risk of severe hypoglycaemia, so the hypoglycaemia plan is part of the prescription. [1] [2]
Child in DKA. Run the protocol: isotonic saline first, insulin at 0.05 to 0.1 unit per kg per hour after volume is restored, no bolus, potassium replaced, dextrose added when glucose falls, and transition to subcutaneous insulin only when ketones clear and the child eats. The most common error is rushing the insulin or the fluid, which is the route to cerebral oedema. [4] [9]
Adolescent with erratic control. Choose a regimen that minimises daily burden — once-daily basal, a pump, or a hybrid closed loop — screen for diabetes distress and disordered eating, give private adolescent time, and build a structured transition to adult care that reconciles the regimen and the devices. Puberty-related insulin resistance often raises requirement, so expect to titrate upward. [1] [11]
Child starting growth hormone. Set the dose, agree the injection routine with the family, monitor IGF-I and growth velocity, and counsel on the long horizon and the safety trio. Growth velocity usually improves in the first year, and a flat response prompts a technique and adherence review before a dose increase. [8] [12]
Sick day on insulin or levothyroxine. The sick-day rule for insulin is never to stop insulin — the child needs more, not less, during illness — and to check glucose and ketones frequently, adjusting the dose and using sick-day rules. Levothyroxine dose is usually unchanged in febrile illness, but absorption and adherence may slip, so the emphasis is on consistent administration rather than a dose change. [1] [4]
Child on a hybrid closed-loop system. The continuous glucose monitor, the pump and the algorithm work together, but the user still sets meal boluses and watches for ketones. The safety issue is pump or infusion-set failure: because there is no subcutaneous basal reserve, ketones rise within hours and DKA can develop fast, so the family needs a ketone plan and a back-up injection regimen. [3] [11]
Complications & Pitfalls
The complications of endocrine prescribing are the toxicities of getting the dose, the timing or the monitoring wrong. The pitfalls are the cognitive errors that get you there. [1]
The classic insulin-prescribing errors are giving an intravenous insulin bolus in DKA, starting insulin before fluid resuscitation, stopping insulin during illness, omitting the basal when transitioning off a drip, and underestimating dehydration. Each is preventable by running the protocol the same way every time. Injecting into lipohypertrophic tissue is the commonest reversible cause of erratic control; detect it by examining the sites and counsel on rotation. An insulin pump interruption causes rapid DKA because there is no basal reserve, so every pump family needs a ketone and pump-failure plan. [4] [9]
The levothyroxine pitfalls are over-replacement, which causes tachycardia, agitation, poor growth and behaviour change, and under-replacement in the first two years, which is a neurodevelopmental emergency. The trap of treating the number rather than the child appears here too: chasing a single TSH rather than the growth and developmental pattern, or changing the dose for a transiently abnormal result. [6] [7]
The growth hormone safety trio is the examiner favourite: intracranial hypertension (headache, visual change, papilloedema), slipped capital femoral epiphysis (limp, hip or knee pain), and glucose intolerance. The pitfall is pushing the IGF-I above the reference range to chase height, which trades a small gain for a real safety cost. [12]
Prognosis & Disposition
The outlook for a child on an endocrine medicine is the outlook for the underlying axis, modified by how promptly and consistently the dose is right. Drug-related crises often recover fully when caught early; chronic under- or over-treatment leaves lasting harm. [1]
The glycemic outcome for a child on insulin depends on regimen fit, technology access, family capacity, adherence and psychosocial context far more than on the choice of analogue. Early good control, measured by HbA1c and time in range, reduces long-term microvascular risk, which is why the first year after diagnosis matters disproportionately. The threshold for higher-acuity care is the child in DKA, the child with severe hypoglycaemia, and the newly diagnosed child needing stabilisation. [2] [11]
The long-term outlook for a child with congenital hypothyroidism treated early and adequately is normal cognition and growth — provided the levothyroxine is given consistently and the dose grows with the child. The trial off therapy for suspected transient disease happens later, never in the first two years. [6]
For a child on growth hormone, the growth response in the first year predicts the trajectory, and the continuation to near-adult height with IGF-I in range is the goal. The follow-up surveillance is 3 to 6 monthly growth and IGF-I, an annual bone age, and a hip and eye review through treatment, with a reassessment for adult deficiency at the end of growth. [8] [12]
The child can be safely managed at home on an endocrine medicine only when the family can reproduce the dose, recognise and treat hypoglycaemia, run a sick-day plan, monitor glucose or attend for TSH or IGF-I, and knows when to seek help. That safety-net is part of the prescription, not an afterthought. [1]
Special Populations
Neonates and infants have immature glucose regulation, milk-fed unpredictability and tiny weight-based doses that make insulin and levothyroxine prescribing especially exact. The first two years are the neurodevelopmental window for thyroid hormone, so the dose, the timing and the recheck schedule are non-negotiable in infancy. [6] [7]
Adolescents carry the heaviest prescribing complexity. Puberty-related insulin resistance can roughly double insulin requirement, adherence slips with risk-taking, disordered eating distorts insulin taking, and driving and alcohol add real safety obligations. The regimen must minimise burden, the visit must include private adolescent time, and the transition to adult care must reconcile the dose, the devices and the monitoring — a documented handover, not an implied one. [1] [11]
Children with coexisting autoimmunity — coeliac disease, thyroiditis, or both — need active screening, because coeliac disease worsens glycaemia and growth and thyroid dysfunction changes insulin requirement. The prescribing decision is never just about the one axis in front of you. [1]
Children with medical complexity and technology dependence are reshaped by enteral feeding, steroids and multiple comorbidities: steroids raise insulin requirement and suppress the growth response, enteral feeding changes carbohydrate timing, and polypharmacy raises interaction risk. The safety-net and the specialist link are tighter here. [8]
Indigenous, remote and rural children face distance from specialist pharmacy and technology, and they more often present late in DKA. Telehealth follow-up, a clear sick-day plan, and a local pathway for emergency supply close some of the gap. [4]
Migrant, refugee and asylum-seeking families may bring language discordance, unfamiliar devices and interrupted supplies, all of which risk a correctly calculated dose being given wrongly or inconsistently at home. A professional interpreter, a taught device, and a written plan in the family's language are the safeguards. [1]
Children from low-income or marginalised groups experience real disparities in access to insulin pumps and continuous glucose monitoring, which feed back into worse glycaemic and quality-of-life outcomes. Prescribing equitably means choosing the regimen that fits the family's real access, and advocating for the funded technology where it is indicated. [11]
Evidence, Guidelines & Regional Differences
The evidence base for paediatric endocrine prescribing is a mix of international consensus guidelines, randomised trials of technology, regional formularies, and consensus on growth hormone. Knowing which to reach for, and which overrides which, is part of the answer. [1]
The ISPAD 2024 Consensus Guidelines are the global reference for insulin, glycemic targets, diabetes technologies and DKA. They set the HbA1c target below 7 percent for most children, endorse basal-bolus, pump and automated insulin delivery regimens, and codify the DKA insulin rate at 0.05 to 0.1 unit per kg per hour with no bolus. The NICE NG18 guideline (UK diabetes in children) and the NICE NG145 thyroid guideline govern UK practice, with BNFc giving the operative paediatric doses. [2] [3] [4]
The randomised evidence for hybrid closed-loop systems in children and adolescents is now strong, showing improved time in range and reduced hypoglycaemia, with quality-of-life benefit extending to historically minoritised youth when access is provided. The ESPE consensus on congenital hypothyroidism (Leger 2014) sets the levothyroxine starting dose and the recheck intervals, and the initial-dose debate — high-dose for rapid TSH normalisation versus lower-dose for fewer over-treatment effects — continues to be examined for cognitive outcome. [5] [6] [7] [11]
For growth hormone, the Growth Hormone Research Society consensus sets the diagnostic and dosing framework, and the GloBE-Reg minimum dataset standardises the monitoring across registries. The funded indications vary by region, and the ongoing controversy over idiopathic short stature — efficacy, cost-effectiveness and the modest height gain — sits alongside the licensed deficiency indications. [8] [12]
The RCH Melbourne Clinical Practice Guidelines and the Australasian Paediatric Endocrine Group drive operative insulin, DKA, levothyroxine and growth hormone protocols. Subsidised access to continuous glucose monitoring and hybrid closed-loop pumps has expanded, and growth hormone is funded through national programmes for licensed indications. [1] [6]
The ISPAD 2024 guidelines and the ESPE congenital hypothyroidism consensus are the global reference frame; the Growth Hormone Research Society consensus sets the growth hormone framework. Always state the guideline you are quoting when you give a drug-specific number. [2] [6] [12]
Exam Pearls
- DKA insulin infusion is 0.05 to 0.1 unit per kg per hour intravenously, started only after intravascular volume is restored; never give an insulin bolus. [4]
- New type 1 diabetes total daily dose is about 0.5 to 0.75 unit per kg per day at diagnosis, lower in the partial-remission honeymoon phase, split roughly half basal and half bolus. [1]
- Basal-bolus regimen: one daily long-acting analogue (glargine, detemir, degludec) plus a rapid-acting analogue (lispro, aspart, glulisine) with each meal. [1]
- ISPAD 2024 glycemic target is HbA1c below 7 percent for most children, individualised, with time-in-range goals alongside capillary glucose. [2]
- Insulin pump interruption causes rapid DKA because there is no subcutaneous basal reserve; ketones rise within hours, so every pump family needs a ketone and failure plan. [3]
- Neonatal congenital hypothyroidism levothyroxine starting dose is 10 to 15 microgram per kg per day once daily, started within the first two weeks to protect neurodevelopment. [6]
- Levothyroxine is given once daily, ideally on an empty stomach, with TSH rechecked at 2 and 4 weeks then every 1 to 3 months in infancy and every 3 to 6 months later. [6]
- Recombinant growth hormone dose is 0.045 to 0.050 mg per kg per day (about 0.15 to 0.17 IU per kg per week) subcutaneous once daily, titrated to IGF-I in the reference range. [8] [12]
- Growth hormone safety surveillance trio: intracranial hypertension (headache, visual change), slipped capital femoral epiphysis (limp), and glucose intolerance. [12]
- Insulin classes by duration: rapid analogues (lispro, aspart, glulisine), soluble (short), isophane or NPH (intermediate), and long-acting analogues (glargine, detemir, degludec). [1]
- Always name the guideline or formulary (ISPAD, ESPE, NICE, BNFc, RCH Melbourne) when you give a drug-specific number, and never quote a remembered paediatric endocrine dose as definitive. [1] [6]
References
- [1]Cengiz E, Danne T, et al International Society for Pediatric and Adolescent Diabetes Clinical Practice Consensus Guidelines 2024: Insulin and Adjunctive Treatments in Children and Adolescents with Diabetes Horm Res Paediatr, 2024.PMID 39884261
- [2]de Bock M, Agwu JC, et al International Society for Pediatric and Adolescent Diabetes Clinical Practice Consensus Guidelines 2024: Glycemic Targets Horm Res Paediatr, 2024.PMID 39701064
- [3]Tauschmann M, Cardona-Hernandez R, et al International Society for Pediatric and Adolescent Diabetes Clinical Practice Consensus Guidelines 2024 Diabetes Technologies: Glucose Monitoring Horm Res Paediatr, 2024.PMID 39884260
- [4]Wolfsdorf JI, Glaser N, et al ISPAD Clinical Practice Consensus Guidelines 2018: Diabetic ketoacidosis and the hyperglycemic hyperosmolar state Pediatr Diabetes, 2018.PMID 29900641
- [5]Brown SA, Kovatchev BP, et al Six-Month Randomized, Multicenter Trial of Closed-Loop Control in Type 1 Diabetes N Engl J Med, 2019.PMID 31618560
- [6]Léger J, Olivieri A, et al European Society for Paediatric Endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism J Clin Endocrinol Metab, 2014.PMID 24446653
- [7]Esposito A, Vigone MC, et al Effect of initial levothyroxine dose on neurodevelopmental and growth outcomes in children with congenital hypothyroidism Front Endocrinol (Lausanne), 2022.PMID 36133316
- [8]Kanakatti Shankar R, Quigley CA, et al Growth and Growth-Promoting Treatments in Turner Syndrome Am J Med Genet C Semin Med Genet, 2025.PMID 39950365
- [9]Azova S, Rapaport R, et al Brain injury in children with diabetic ketoacidosis: Review of the literature and a proposed pathophysiologic pathway for the development of cerebral edema Pediatr Diabetes, 2021.PMID 33197066
- [10]Brown KM, Glaser NS, et al Rehydration Rates and Outcomes in Overweight Children With Diabetic Ketoacidosis Pediatrics, 2023.PMID 37920947
- [11]Marks BE, Grundman JB, et al Hybrid Closed Loop Systems Improve Glycemic Control and Quality of Life in Historically Minoritized Youth with Diabetes Diabetes Technol Ther, 2024.PMID 38444316
- [12]Chen SC, Bryce J, et al Development of a Minimum Dataset for the Monitoring of Recombinant Human Growth Hormone Therapy in Children with Growth Hormone Deficiency: A GloBE-Reg Initiative Horm Res Paediatr, 2024.PMID 37703843