Paeds · endocrinology-diabetes-and-growth
Adrenal insufficiency and adrenal crisis
Also known as Adrenal insufficiency · Addison disease · Adrenal crisis · Primary adrenal insufficiency · Secondary adrenal insufficiency · Adrenal failure · Glucocorticoid deficiency
Fellowship guide to adrenal insufficiency and adrenal crisis in children: the primary-versus-secondary split, autoimmune Addison disease and the salt-wasting crisis, glucocorticoid-withdrawal secondary insufficiency, the cortisol-ACTH-renin work-up, acute resuscitation with fluids and parenteral hydrocortisone, and lifelong replacement with a stress-dose plan.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single idea that organises everything is where the axis breaks. Break it at the adrenal cortex and the child loses cortisol and aldosterone, so ACTH and renin climb and potassium rises. Break it at the pituitary or hypothalamus, or suppress it with exogenous steroid, and the child loses cortisol alone, so aldosterone, renin and potassium stay near-normal. That is why one biochemical disease presents as two different emergencies. [2]
This page covers the recognition and management of adrenal insufficiency and adrenal crisis in children: primary versus secondary disease, the autoimmune Addison picture, glucocorticoid-induced suppression, the salt-wasting crisis, the cortisol-ACTH-renin work-up, acute resuscitation with fluids and parenteral hydrocortisone, lifelong hydrocortisone and fludrocortisone replacement, and the stress-dose and emergency-injection plan. It links to the congenital adrenal hyperplasia and hypopituitarism leaves rather than repeating their full pathways. [1]
Overview & Definition
A child first seen pale, drowsy and vomiting with low blood pressure is the opening scene of adrenal insufficiency. The condition is a deficiency of adrenal glucocorticoid (cortisol), with or without mineralocorticoid (aldosterone), arising from disease of the adrenal cortex itself or from loss of the trophic drive that keeps it working. [2]
When the cortex fails directly the disease is primary, and both cortisol and aldosterone fall. The loss of aldosterone is what makes primary disease biochemically loud: the child wastes sodium, retains potassium, and dehydrates. ACTH rises unchecked and drives the hyperpigmentation that is the physical signature of chronic primary disease. This is Addison disease when autoimmune, and congenital adrenal hyperplasia when the cause is an enzyme block. [1]
When the trophic drive from the pituitary or hypothalamus fails, the disease is secondary or tertiary, and only cortisol falls. Aldosterone is controlled largely by the renin-angiotensin system rather than by ACTH, so sodium, potassium and blood pressure hold up far better. By far the commonest cause in children is suppression of the axis by exogenous glucocorticoid, given as oral, inhaled, nasal or topical steroid that is then withdrawn. [6]
Classification
The fastest way to classify adrenal insufficiency is by the level of the break, because level decides the electrolytes, the physical signs, and the long-term drugs. The figure below splits the axis into primary and secondary-tertiary and tabulates the commonest paediatric cause of each. [1]
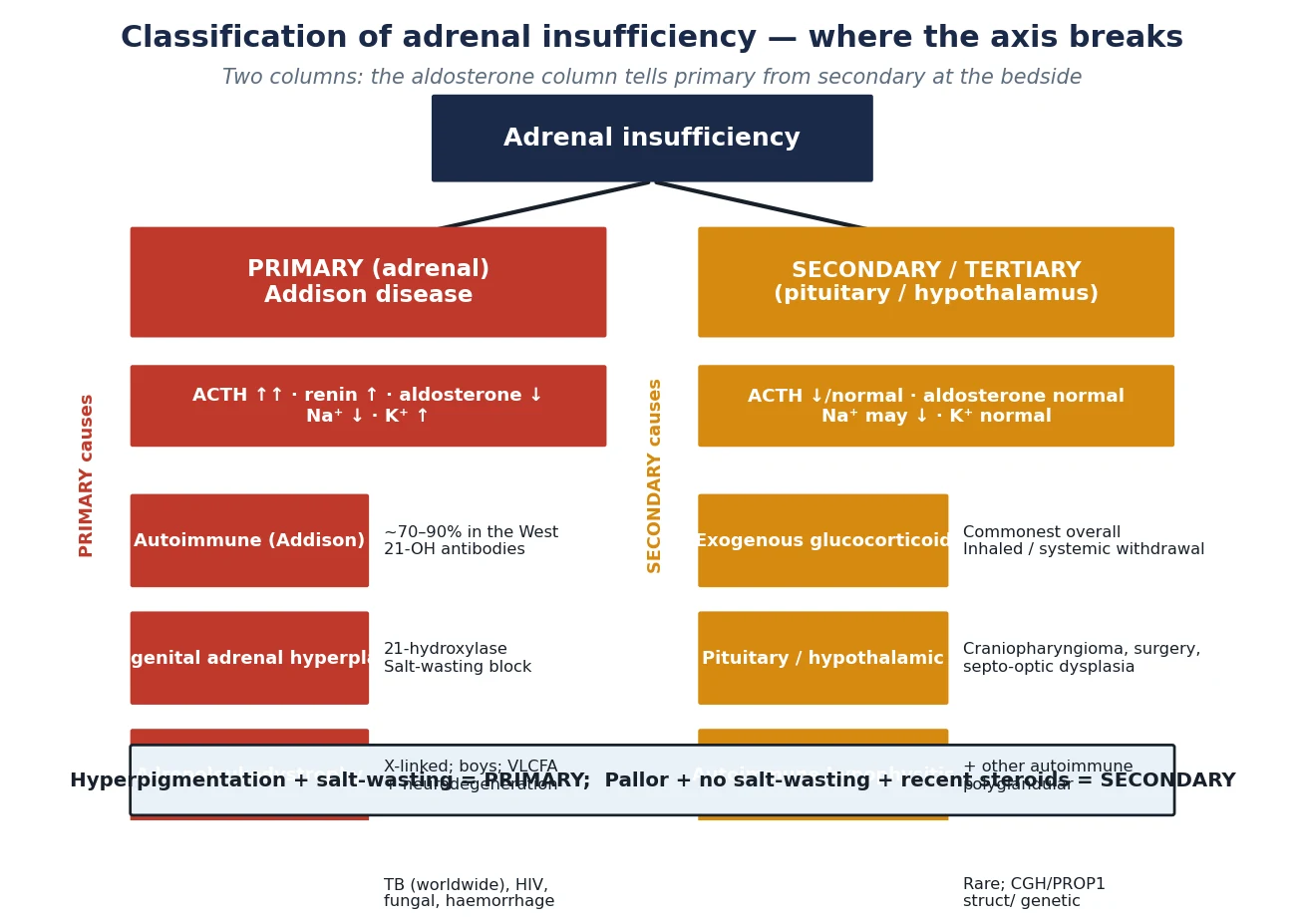
Primary (Addison)
- Adrenal cortex fails: cortisol AND aldosterone low
- ACTH high, renin high, potassium high, sodium low
- Autoimmune in 70–90% of Western cases (21-OH antibodies)
- Hyperpigmentation, salt-craving, weight loss; crisis with shock
Secondary / Tertiary
- Pituitary/hypothalamus fails: cortisol low alone
- ACTH low, aldosterone normal, potassium normal
- Commonest cause is glucocorticoid withdrawal (inhaled/systemic)
- No hyperpigmentation; crisis less salt-wasting but still hypoglycaemic
Within primary disease the autoimmune form dominates in the developed world, driven by 21-hydroxylase antibodies and often clustering with other autoimmune endocrinopathies in the autoimmune polyglandular syndromes. In tuberculous-endemic regions tuberculosis overtakes autoimmunity, and fungal infection, HIV, infiltrative disease and bilateral adrenal haemorrhage fill in the rest. In boys with primary insufficiency and neurological decline, adrenoleukodystrophy — an X-linked peroxisomal defect that accumulates very-long-chain fatty acids — is the diagnosis not to miss. [2] [7]
Secondary disease is overwhelmingly iatrogenic. Any child who has taken supraphysiological glucocorticoid for more than two to four weeks has a suppressed axis, and the suppression can persist for many months after the drug stops. The other causes — pituitary and hypothalamic tumours, craniopharyngioma and its surgery, septo-optic dysplasia, and congenital hypopituitarism — are rarer but carry the same risk of crisis under stress. [6]
Epidemiology & Risk Factors
Autoimmune Addison disease is uncommon in children but not rare: population estimates place the prevalence of primary adrenal insufficiency in the low hundreds per million, with a female predominance and a peak diagnosis in the third and fourth decades, so the paediatric endocrinologist meets it in adolescents and in the siblings of affected families. Autoimmune clustering is the strongest risk marker, so a child with type 1 diabetes and thyroiditis who develops fatigue and pigmentation has Addison until tested. [7] [10]
Adrenal crisis, by contrast, is common in anyone with known insufficiency who meets a stressor. Registry and case-control data show that roughly one in twenty patients with chronic adrenal insufficiency has a crisis each year, and that infections, gastrointestinal illness with vomiting, surgery, and missed or under-dosed replacement are the recurring precipitants. Children are over-represented because intercurrent febrile illness is frequent and because adherence to a thrice-daily tablet is hard. [8] [9]
CRISIS
Primary loses cortisol and aldosterone; secondary loses cortisol alone
High renin and high potassium point to primary (aldosterone-deficient) disease
Fever, gastroenteritis, surgery, and missed doses precipitate crisis
Withdrawal of inhaled or systemic steroid is the commonest secondary cause
Parent-held emergency hydrocortisone is the single best preventive tool
Double or triple the oral dose for illness; parenteral if vomiting
Pathophysiology
Cortisol is the stress hormone that holds blood pressure, glucose and the immune system together, and aldosterone holds sodium, potassium and circulating volume. Both are made in the adrenal cortex under the trophic drive of the hypothalamic-pituitary axis, and the figure below shows what the body loses when each part of that axis fails. [2]
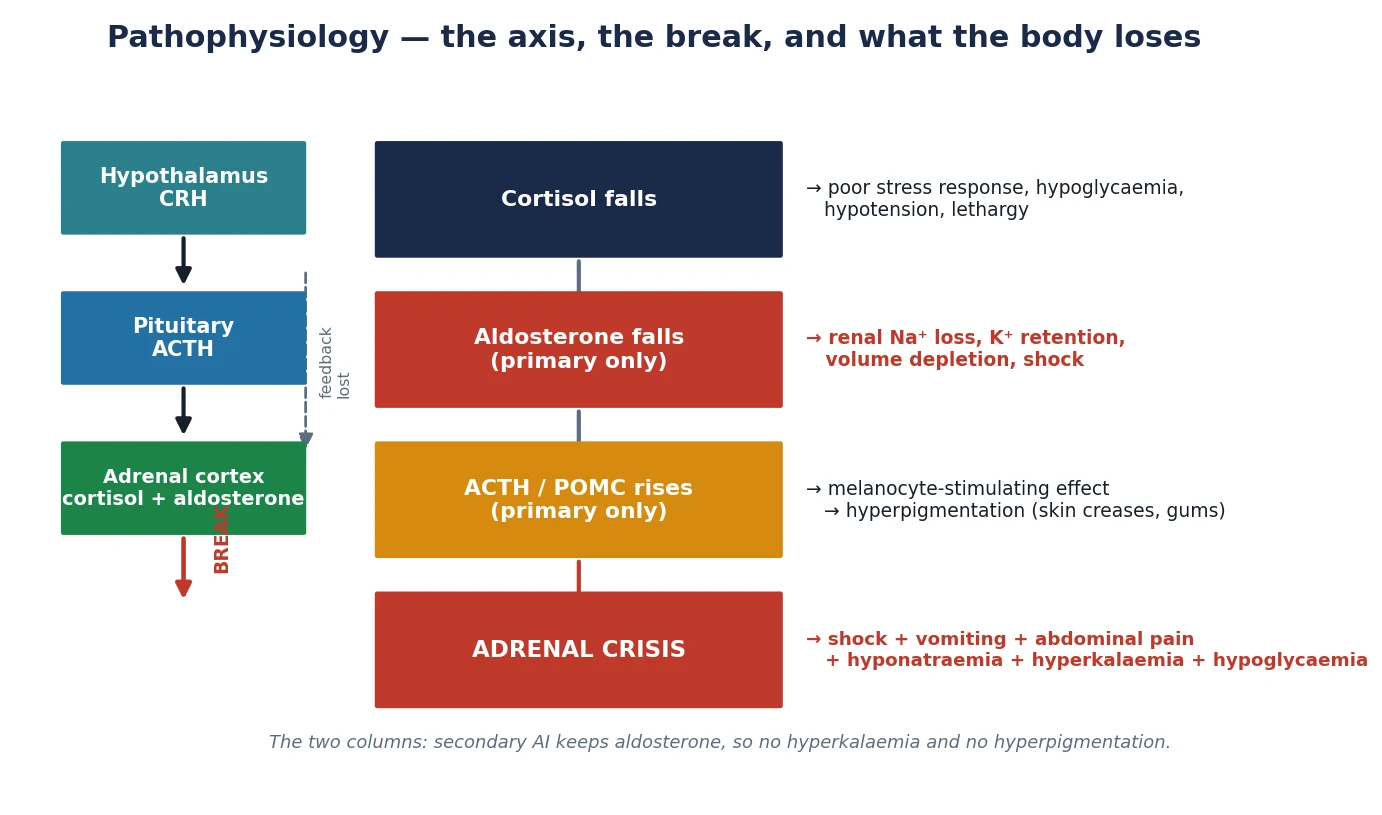
When cortisol falls, negative feedback on the pituitary is lost, so ACTH climbs. In primary disease the climbing ACTH cannot raise cortisol, but it drives the co-secreted melanocyte-stimulating fragments of the POMC precursor, and those stain the skin — hyperpigmentation of the palmar creases, gum margins, areolae, old scars and sun-exposed areas is the visible clue to chronic primary disease. Secondary disease, with its low ACTH, never pigments; instead the child looks pale. [1] [2]
Aldosterone is the other half of the story, and it is the half that explains the electrolytes. Aldosterone secretion is governed mainly by the renin-angiotensin system, not by ACTH, so it survives secondary adrenal failure but collapses in primary disease. The child with primary insufficiency wastes sodium and water, retains potassium, and depletes the circulating volume until shock supervenes. The child with secondary insufficiency keeps aldosterone, so potassium stays normal and the mild hyponatraemia, when present, comes from inappropriate antidiuretic hormone secretion rather than from salt loss. [2]
Cortisol deficiency itself does three things that bring the child to hospital: it permits hypoglycaemia by removing gluconeogenesis, it permits hypotension by removing vascular catecholamine sensitisation, and it removes the anti-inflammatory brake. Add an intercurrent infection and the cortisol-deficient child cannot mount the stress response, so what should be a febrile illness becomes a collapse. That decompensation is adrenal crisis. [4]
Clinical Presentation
The presentation of adrenal insufficiency is either chronic and creeping or acute and catastrophic, and a child can move from one to the other over hours. The chronic picture is fatigue, weight loss, anorexia, nausea, abdominal pain, salt-craving, dizziness on standing, and — in primary disease — progressive hyperpigmentation and postural hypotension. Parents describe a child who has "gone brown" and is drinking salt water or eating salt. [2] [7]
Acute adrenal crisis declares itself with the triad of hypotension refractory to fluids, gastrointestinal symptoms, and the biochemical pattern of hyponatraemia, hyperkalaemia and hypoglycaemia. In children the hypoglycaemia is often the dominant feature and may present as seizures or reduced consciousness before shock is obvious. Abdominal pain can be severe enough to mimic an acute abdomen, and fever is common either from the precipitating infection or from the cortisol deficiency itself. [3] [4]
The child with secondary insufficiency presents a little differently. The hyperpigmentation and the hyperkalaemia are absent, the crisis is less salt-wasting, but the hypoglycaemia and the collapse under stress are just as dangerous. The clue is in the history: recent or current glucocorticoid therapy, a pituitary lesion or its surgery, or a known syndrome such as septo-optic dysplasia. A child who has just stopped a long steroid course and becomes unwell is cortisol-deficient until proven otherwise. [6]
Differential Diagnosis
The differential of an adrenal crisis is the differential of the sick hyponatraemic, hypoglycaemic child, and sepsis sits at the top of every list. The discriminating features are the electrolyte pattern and the response to treatment: a child with gastroenteritis is rarely both hyponatraemic and hyperkalaemic, and a child whose shock corrects only after hydrocortisone is given was cortisol-deficient. Cover sepsis regardless, because sepsis and adrenal crisis coexist and are indistinguishable at the bedside. [3] [4]
For the chronic presentation the differential is the tired, losing-weight child: malignancy, inflammatory bowel disease, chronic infection, anorexia nervosa, and occult coeliac disease all overlap with the fatigue and weight loss of Addison disease. The discriminator is the electrolyte and pigmentation axis: a child who is pigmented and hyponatraemic with a high potassium is not depressed or anorexic. Postural hypotension with weight loss in an adolescent is Addison disease until a morning cortisol is checked. [2] [7]
Hyponatraemia with hyperkalaemia has a short, high-yield list: primary adrenal insufficiency, congenital adrenal hyperplasia, renal salt-wasting, pseudohypoaldosteronism, and severe renal failure. The blood pressure sorts them — the adrenal child is hypotensive, the renal child often hypertensive — and the glucose and the ACTH close the gap. Do not accept "gastroenteritis" as the explanation for a high potassium in a sick child. [1]
Clinical & Bedside Assessment
The bedside assessment runs on two tracks at once: judge how sick the child is now, and gather the clues that localise the break. Start with the airway, breathing and circulation, because the crisis is a volume-depleted shock state. Capillary refill, heart rate, blood pressure and conscious level, with a bedside glucose, tell you whether you are resuscitating or investigating first. [4]
The focused examination looks for the stigmata of chronic cortisol and aldosterone loss. Look at the skin for hyperpigmentation — emphasised in the palmar creases, knuckles, gum margins, areolae, recent scars and sun-exposed areas — and for vitiligo, which marks the autoimmune diathesis. Look for postural drop in the blood pressure, for cachexia, and for the signs of an associated autoimmune disease such as a goitre or diabetic ketosis. Plot the weight, because weight loss is the long-term trend that flags chronic disease. [1] [7]
Measure the blood pressure in all four limbs if you suspect a secondary cause, because a pituitary lesion may sit with other congenital or vascular signs. Examine the visual fields and the fundi, look for midline defects that suggest septo-optic dysplasia, and ask about growth — a child with combined pituitary deficiency may also have growth-hormone failure and short stature. In a boy with primary insufficiency and neurological or behavioural change, examine for the spasticity, visual loss and deafness of adrenoleukodystrophy. [2] [6]
Investigations
The laboratory plan answers two questions at once: is the cortisol low, and where is the break. Draw the first set of bloods before you give hydrocortisone if you can, because one cortisol and one ACTH measured in the acute moment often settle the diagnosis; never delay treatment to wait for them. [1]
A morning cortisol is the screening workhorse. A random stress cortisol above 500 nanomoles per litre in a sick child effectively excludes adrenal insufficiency, while a low morning cortisol with a high ACTH confirms primary disease and a low cortisol with a low or inappropriately normal ACTH confirms secondary disease. Renin and aldosterone close the loop: high renin with low aldosterone is the primary pattern. Check the urea, electrolytes, glucose and a venous gas in every crisis, because the electrolyte and acid-base pattern is part of the diagnosis. [1] [5]
The 250-microgram ACTH (cosyntropin) stimulation test is the confirmatory standard for primary disease: a cortisol that fails to rise above 500 nanomoles per litre at 30 or 60 minutes confirms adrenal failure. For suspected secondary disease a low-dose or insulin-tolerance test is more sensitive, because the chronically under-stimulated adrenal can still respond to a pharmacological dose. In confirmed primary disease, measure 21-hydroxylase antibodies to prove autoimmunity, image the adrenals with computed tomography or ultrasound to find infiltrative, infective or haemorrhagic causes, and in any boy send very-long-chain fatty acids to exclude adrenoleukodystrophy. [1] [5]
Management — Resuscitation
Adrenal crisis is a volume-depleted, cortisol-deficient shock state, and it kills if untreated. Resuscitation runs on three tracks at once: restore the circulating volume, replace the missing cortisol, and correct the glucose and electrolytes while you treat the precipitant. [1] [4]
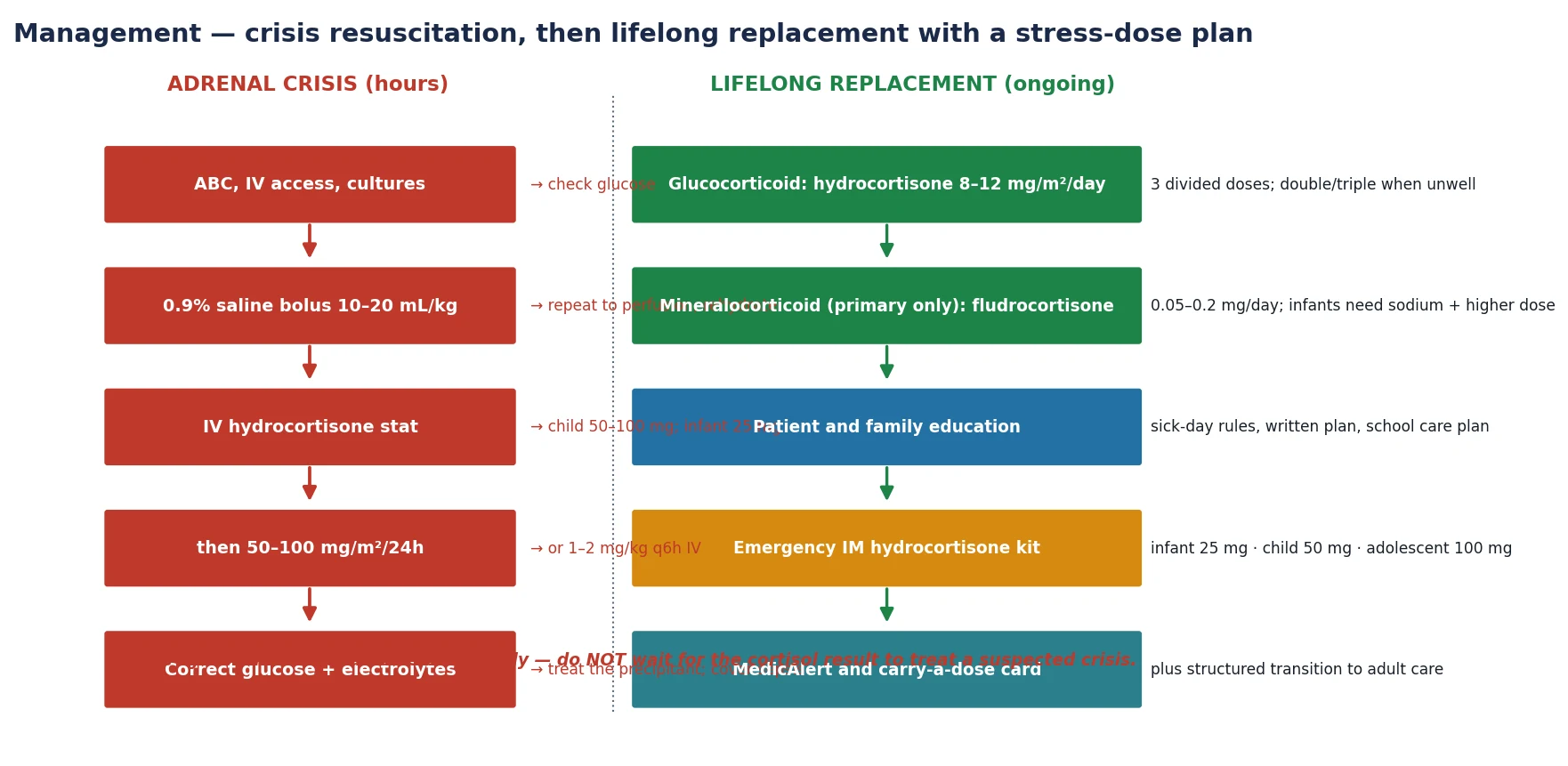
Give an isotonic fluid bolus of 10 to 20 millilitres per kilogram of 0.9% saline and repeat to perfusion, then continue rehydration over the next day. Give parenteral hydrocortisone immediately — 50 to 100 milligrams intravenously for a child, and 25 milligrams for an infant or neonate — then continue as 50 to 100 milligrams per square metre per 24 hours, either by continuous infusion or divided every six hours. Correct the hypoglycaemia with intravenous dextrose, and treat hyperkalaemia expectantly because it usually settles as volume, cortisol and aldosterone activity return. [1] [4]
Across the Endocrine Society (Bornstein 2016), the European consensus (Husebye 2014) and the 2024 joint glucocorticoid-induced guideline, the acute crisis is treated with isotonic volume, parenteral hydrocortisone at stress doses, glucose correction, and cover for the precipitant. Local protocols differ on whether hydrocortisone is given by continuous infusion or by divided bolus, but the principle — fluids, hydrocortisone, electrolyte and glucose correction in parallel — is universal. Empiric treatment must never wait for a cortisol result. [1] [5]
Cover sepsis with cultures and empiric antibiotics, because sepsis and adrenal crisis are clinically indistinguishable and often coexist. Identify and treat the precipitant — an infection, a missed dose, surgery or a gastrointestinal illness. Escalate to a paediatric intensive care setting if shock persists after the first fluid bolus and steroid dose, because the refractory child may need vasoactive support while the cortisol takes hold. [3] [4]
Management — Definitive & Stepwise
Once the child is stable, definitive management is lifelong hormone replacement built around a stress-dose and emergency-injection plan. The aim is to replace what is missing, mimic the physiological cortisol rhythm, and keep the child safe under illness and surgery — all without the growth suppression and iatrogenic Cushing that come from over-treatment. [1]
The definitive pathway, from crisis to lifelong plan
Resuscitate: fluids, parenteral hydrocortisone, glucose and electrolytes
Confirm the diagnosis and localise the break: cortisol, ACTH, renin, aldosterone, antibodies, imaging
Start oral hydrocortisone 8–12 mg/m²/day in three divided doses (primary and secondary)
Add fludrocortisone 0.05–0.2 mg/day for primary disease only, with infant sodium supplements
Build the stress-dose plan: double or triple oral hydrocortisone for illness, parenteral if vomiting
Issue a parent-held emergency intramuscular hydrocortisone kit and teach the family to use it
Add a MedicAlert, a school care plan, and structured transition to adult endocrinology
Replace glucocorticoid with hydrocortisone at 8 to 12 milligrams per square metre per day in three divided doses, with a larger morning dose to mimic the cortisol rhythm. Reserve longer-acting steroids such as prednisolone and dexamethasone for older adolescents or specific suppression, because they suppress growth more than hydrocortisone. In primary disease add fludrocortisone at 0.05 to 0.2 milligrams per day; infants need a higher relative dose and oral sodium supplementation because their renal sodium handling is immature. Secondary disease does not need fludrocortisone, because aldosterone is intact. [1] [5]
Replacement and stress doses in children with adrenal insufficiency
The stress-dose principle is the part that saves lives between clinics. The adrenal child cannot mount a cortisol rise under illness, so the family must: double or triple the oral hydrocortisone for any febrile illness or significant stress, give the parenteral intramuscular dose if the child is vomiting or severely unwell, and present to hospital early. Teach the family to give the emergency injection first and call for help second, because a delay of hours in a vomiting child is the gap in which crisis deaths happen. [9] [8]
Monitoring is clinical more than biochemical. Plot the growth — height velocity falling suggests over-replacement, and weight and puberty advancing too quickly suggests under-replacement — and check the renin periodically in primary disease to titrate fludrocortisone. Chasing a single cortisol target misses the point; the child who is growing well, energised and crisis-free is well replaced. [1] [5]
Specific Subtypes & Scenarios
The scenarios below are the ones that present, collapse, and are misdiagnosed, so they are the highest-yield material for the bedside and the written paper. [3]
The child with undiagnosed autoimmune Addison disease is the classic miss. The presentation is months of fatigue, weight loss, abdominal pain and progressive pigmentation, finally tipping into crisis with a chest or gastrointestinal infection. The diagnosis is delayed because each symptom is attributed to something else — school stress, anorexia, viral illness — until the electrolyte pattern and the high ACTH make it obvious. A morning cortisol in any pigmented, losing-weight adolescent closes the gap. [2] [7]
The steroid-withdrawn child is the commonest secondary scenario. A child treated for asthma, nephrotic syndrome, juvenile arthritis or inflammatory bowel disease reduces or stops a long glucocorticoid course, then meets an infection and collapses. The crisis is less salt-wasting but the hypoglycaemia and hypotension are real, and the history of recent steroid is the clue. Never assume the axis has recovered within months of stopping a long course. [6]
The boy with adrenoleukodystrophy is the diagnosis not to miss in primary insufficiency with neurological signs. X-linked adrenoleukodystrophy causes primary adrenal failure alongside progressive demyelination, spasticity, visual and hearing loss, and behavioural change. Any boy with primary adrenal insufficiency should have very-long-chain fatty acids measured, because the adrenal failure can precede the neurological disease by years and bone-marrow or gene therapy is time-critical. [2]
The neonate or infant with bilateral adrenal haemorrhage is the acute surgical-look-alike. Birth trauma, perinatal asphyxia, severe sepsis (Waterhouse-Friderichsen from meningococcaemia), and anticoagulation or antiphospholipid in the neonate can all haemorrhage both adrenals and present as an acute abdomen with shock and a falling haemoglobin. The clue is the precipitant, and imaging shows the enlarged, haemorrhagic adrenals. [3]
Complications & Pitfalls
The complications divide into those of the untreated disease and those of its treatment. Untreated or under-treated insufficiency brings adrenal crisis — the lethal event — and, in primary disease, the long-term toll of chronic salt-wasting, hypoglycaemia, growth failure and the autoimmune clustering that brings thyroid, diabetes and gonadal failure. Over-treated insufficiency brings iatrogenic Cushing, growth suppression and osteopenia from chronic glucocorticoid excess. [1] [7]
The lethal pitfall is the missed crisis. The child treated as gastroenteritis or sepsis while the adrenal fails is the preventable death, and the countermeasure is a low threshold to give empiric hydrocortisone and fluids in any sick child with an unexplained electrolyte pattern or shock refractory to fluid. One dose of intravenous hydrocortisone is harmless if you are wrong and life-saving if you are right. [3] [4]
Risk of adrenal crisis per year in chronic adrenal insufficiency
high
Roughly 5–10% of adults with chronic AI experience a crisis each year; children carry higher risk because intercurrent illness is frequent and adherence to thrice-daily dosing is hard. A written sick-day plan and a parent-held emergency injection reduce crisis and death.
The treatment pitfalls are concrete. Forgetting the stress dose during illness or surgery allows crisis in a treated child. Stopping fludrocortisone in primary disease allows salt-wasting. Chasing a single cortisol number instead of the whole child leads to over- or under-treatment. And assuming the axis has recovered within months of steroid withdrawal allows a crisis at the next infection. [6] [8]
Prognosis & Disposition
With early diagnosis, disciplined replacement and a working stress-dose plan, the outlook for a child with adrenal insufficiency is good. Intellectual development is normal, growth and puberty proceed normally if the glucocorticoid dose is well balanced, and fertility and lifespan approach the general population. The single biggest modifiable factor is whether the diagnosis is made and the emergency plan is built before a crisis. [1] [7]
The long-term battleground is the balance between under- and over-treatment. Under-treatment permits crises, salt-wasting and the androgen-driven effects of rising ACTH in primary disease; over-treatment suppresses growth and bone health. Final height and bone density are the two outcomes that track the quality of replacement over years, and they reward the clinician who titrates to the child rather than to the number. [5]
Disposition is lifelong. The child needs a named endocrinologist, a primary-care partner who knows the stress-dose rules, a MedicAlert, a school care plan, and a structured transition to adult endocrinology in late adolescence. The crisis-prevention literature shows that patient education and the emergency injection are the interventions that reduce crisis frequency and mortality, so the time spent teaching the family is the highest-yield clinical act. [8] [9]
Special Populations
The adolescent at transition carries the cumulative burden of a chronic, invisible disease into independence. The challenges are adherence to a thrice-daily tablet, the carry-over of a parent-managed emergency plan into self-management, the risk-taking and alcohol exposure that can precipitate crisis, and the reproductive and bone-health questions that attend chronic glucocorticoid replacement. A structured, honest transition that rehearses the sick-day rules and the emergency injection is the standard of care. [5]
The child with an autoimmune polyglandular syndrome is the one whose adrenal insufficiency will not travel alone. Type 1 autoimmune polyglandular failure (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, AIRE gene) brings chronic mucocutaneous candidiasis and hypoparathyroidism in childhood; type 2 brings type 1 diabetes and autoimmune thyroid disease. A child diagnosed with Addison disease should be screened for the associated endocrinopathies, and a child with type 1 diabetes who develops fatigue and pigmentation has Addison until tested. [7] [10]
In Australia and Aotearoa New Zealand, autoimmune Addison disease is the dominant primary cause and tuberculosis is rare, but the picture differs in migrant and refugee communities where tuberculous and infective adrenalitis are more common. Specialist care is delivered through regional paediatric endocrinology networks with retrieval for crisis, and the stress-dose and emergency-injection education is standard. Equity of access to a hydrocortisone emergency kit and to culturally safe education is a determinant of survival in remote and Indigenous communities. [1]
The child in a resource-limited or remote setting faces both a higher infective burden and the logistics of accessing hydrocortisone and fludrocortisone. Tuberculous adrenalitis remains a major cause of primary insufficiency in endemic regions, and the absence of a reliable emergency injection makes crisis more lethal. Advocacy for an emergency kit, for school-based education, and for a reliable supply of replacement is part of the medical care. [2] [3]
Evidence, Guidelines & Regional Differences
The evidence base rests on the Endocrine Society guidelines and a long registry and case-control tradition. The 2016 Endocrine Society guideline on primary adrenal insufficiency (Bornstein) set the modern standard for diagnosis, hydrocortisone and fludrocortisone replacement, stress dosing and crisis prevention, and the 2014 European consensus (Husebye) extended it with operational detail. The 2024 European Society of Endocrinology and Endocrine Society joint guideline addressed glucocorticoid-induced adrenal insufficiency, the commonest secondary form. [1] [5] [6]
The adrenal-crisis literature has matured into an epidemiology of its own. Hahner established the incidence and the preventable burden of crisis in chronic insufficiency, the recent case-control work (Chifu 2023) named the recurring precipitants, and the 2019 New England Journal of Medicine review (Rushworth and Falhammar) codified the acute resuscitation. The Lancet 2026 Addison disease review (Dong and Tomlinson) is the current synthesis of the primary disease. [4] [8] [9] [7]
The 2007 Pediatrics statement (Shulman and the Lawson Wilkins committee) remains the paediatric anchor: adrenal insufficiency is still a cause of morbidity and death in childhood, and the preventable failures are the missed diagnosis and the unbuilt stress-dose plan. That framing — that children still die of this treatable disease — is the reason every candidate must know the empiric hydrocortisone dose. [3]
The remaining controversies are practical: the glucocorticoid preparation and regimen that best mimic physiology without suppressing growth, the role of modified-release hydrocortisone, the threshold for screening the at-risk child with type 1 diabetes, and the optimal model of patient education to prevent crisis. A fellowship candidate names the debate and the standard, not a false consensus. [5] [6]
Exam Pearls
One-sentence answer: the approach to suspected adrenal crisis
A child in shock unresponsive to fluids, or with unexplained hyponatraemia, hyperkalaemia and hypoglycaemia, has adrenal crisis until proven otherwise: resuscitate with 0.9% saline boluses and parenteral hydrocortisone (child 50 to 100 mg, infant 25 mg), check the cortisol, ACTH, renin, aldosterone and glucose before or alongside the first dose, then replace hydrocortisone and fludrocortisone with a stress-dose and emergency-injection plan.
The split
- Primary = adrenal failure: cortisol AND aldosterone low; ACTH high; K+ high; hyperpigmentation
- Secondary = pituitary/steroid: cortisol low alone; ACTH low; K+ normal; no pigmentation
- Aldosterone is controlled by renin-angiotensin, NOT ACTH — that is why it survives secondary disease
- Commonest secondary cause in children is glucocorticoid withdrawal
The crisis
- Shock unresponsive to fluids + vomiting + abdominal pain = adrenal crisis until proven otherwise
- Hyponatraemia + hyperkalaemia + hypoglycaemia in a sick child = primary AI
- Treat empirically: 0.9% saline 10–20 mL/kg bolus + IV hydrocortisone BEFORE the cortisol result
- Cover sepsis — it coexists and is indistinguishable
Doses to know
- Acute hydrocortisone: infant/neonate 25 mg, child 50–100 mg IV stat, then 50–100 mg/m²/24h
- Maintenance hydrocortisone: 8–12 mg/m²/day in 3 divided doses
- Fludrocortisone (primary only): 0.05–0.2 mg/day; infants need sodium
- Stress: 2–3× oral for illness; parenteral if vomiting; emergency IM kit
Frequently misremembered facts, stated correctly: secondary adrenal insufficiency does NOT cause hyperkalaemia, because aldosterone is intact; a random stress cortisol above 500 nanomoles per litre effectively excludes adrenal insufficiency; one dose of empiric intravenous hydrocortisone is harmless if wrong and life-saving if right; and a child who has stopped a long steroid course can still collapse months later. [4] [6]
The lesion-to-sign pairings are the fastest route to a bedside answer and the highest-yield material for a written or oral question: hyperpigmentation with hyperkalaemia is primary (Addison); a pale child with a normal potassium and a recent steroid history is secondary; shock unresponsive to fluids with hypoglycaemia is adrenal crisis; and a boy with primary insufficiency and neurological decline is adrenoleukodystrophy until the very-long-chain fatty acids come back. [2] [3]
References
- [1]Bornstein SR; Allolio B; Arlt W; et al Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2016.PMID 26760044
- [2]Charmandari E; Nicolaides NC; Chrousos GP Adrenal insufficiency. Lancet, 2014.PMID 24503135
- [3]Shulman DI; Palmert MR; Kemp SF; Lawson Wilkins Drug and Therapeutics Committee Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics, 2007.PMID 17242136
- [4]Rushworth RL; Torpy DJ; Falhammar H Adrenal Crisis. N Engl J Med, 2019.PMID 31461595
- [5]Husebye ES; Allolio B; Arlt W; et al Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med, 2014.PMID 24330030
- [6]Beuschlein F; Dekkers OM; Arlt W; et al European Society of Endocrinology and Endocrine Society Joint Clinical Guideline: Diagnosis and Therapy of Glucocorticoid-induced Adrenal Insufficiency. J Clin Endocrinol Metab, 2024.PMID 38724043
- [7]Dong VH; Husebye ES; Tomlinson JW; et al Clinical features, investigation, and management of Addison's disease. Lancet Diabetes Endocrinol, 2026.PMID 41587556
- [8]Chifu I; Quinkler M; Hahner S; et al Predisposing factors for adrenal crisis in chronic adrenal insufficiency: a case-control study. Eur J Endocrinol, 2023.PMID 38006230
- [9]Hahner S; Loeffler M; Bleicken B; et al Epidemiology of adrenal crisis in chronic adrenal insufficiency: the need for new prevention strategies. Eur J Endocrinol, 2010.PMID 19955259
- [10]Pearce SHS; Napier CM; Mitchell AL; et al MANAGEMENT OF ENDOCRINE DISEASE: Residual adrenal function in Addison's disease. Eur J Endocrinol, 2021.PMID 33306039