Paeds · endocrinology-diabetes-and-growth
Congenital adrenal hyperplasia
Also known as Congenital adrenal hyperplasia · 21-hydroxylase deficiency · CAH · Adrenogenital syndrome · Salt-wasting congenital adrenal hyperplasia · Classic 21-hydroxylase deficiency
Fellowship guide to congenital adrenal hyperplasia: the cortisol-androgen split, the 21-hydroxylase block, the salt-wasting crisis versus virilisation, the 17-OHP screen and confirmatory work-up, and lifelong hydrocortisone and fludrocortisone replacement with stress dosing.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single fact that organises everything in congenital adrenal hyperplasia is the enzyme block. Stop cortisol production and the pituitary loses its negative feedback, so ACTH climbs and the adrenal cortex hyperplasia. The precursors that cannot pass the block pile up behind it and spill into the androgen pathway. That is why one genetic disease shows up as a salt-losing crisis, an endocrine emergency, and a disorder of sex development all at once. [2] [3]
This page covers the recognition and management of congenital adrenal hyperplasia: the clinical forms, the steroidogenesis block, the salt-wasting crisis, ambiguous genitalia, the 17-hydroxyprogesterone newborn screen and its pitfalls, the confirmatory work-up, acute resuscitation, lifelong glucocorticoid and mineralocorticoid replacement, and family counselling. It links to the dedicated leaves for disorders of sex development and adrenal insufficiency rather than repeating their full pathways. [1]
Overview & Definition
Congenital adrenal hyperplasia is a family of autosomal recessive enzyme defects of adrenal steroidogenesis. More than nine in ten cases are 21-hydroxylase deficiency caused by mutations in CYP21A2, and the rest are rarer defects such as 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase, 17α-hydroxylase and the StAR cholesterol-transport defect. The unifying mechanism is partial or complete loss of an enzyme that the adrenal cortex needs to make cortisol, with a knock-on rise in ACTH and a build-up of upstream precursors. [2]
The clinically useful distinction is between classic and non-classic disease. Classic 21-hydroxylase deficiency has little or no enzyme activity and presents in the newborn period — most often with salt-wasting, sometimes with virilisation alone. Non-classic disease retains enough activity to escape the newborn period and declares itself later with signs of androgen excess. The distinction matters at the bedside because the classic salt-wasting form is a time-critical neonatal emergency. [1] [7]
Classification
The fastest way to classify congenital adrenal hyperplasia is by clinical severity, because severity dictates how urgently the child presents and how aggressively they must be replaced. The figure below splits 21-hydroxylase deficiency into its three clinical forms, then tabulates the rarer enzyme defects by what each does to cortisol, aldosterone, androgens and blood pressure. [1]

Classic salt-wasting
- ~75% of classic 21-OHD; near-zero enzyme activity
- No cortisol AND no aldosterone
- Neonatal salt-wasting crisis at 1–3 weeks
- 46,XX: virilised external genitalia; 46,XY: no genital clue
Classic simple-virilising
- ~25% of classic 21-OHD; enough aldosterone spared
- Low cortisol + androgen excess, no overt salt-wasting
- 46,XX: virilised genitalia may be the only clue
- Still stress-dose dependent
Non-classic (late-onset)
- Partial activity; escapes the newborn period
- Childhood: precocious pubarche, rapid growth, advanced bone age
- Adolescent: hirsutism, acne, oligomenorrhoea, subfertility
- Often the answer in a PCOS-look-alike
The rarer defects have a signature worth remembering. 11β-hydroxylase deficiency accumulates 11-deoxycorticosterone, a mineralocorticoid, so the child is hypertensive rather than salt-wasting but still virilised. 17α-hydroxylase deficiency also causes hypertension and, because it blocks androgen synthesis, undervirilises a 46,XY infant and causes sexual infantilism. 3β-hydroxysteroid dehydrogenase deficiency and the StAR cholesterol-transport defect both salt-waste and undervirilise. The hypertension-versus-salt-wasting and virilise-versus-undervirilise axes do most of the diagnostic work when 21-hydroxylase is excluded. [2] [3]
Epidemiology & Risk Factors
Classic 21-hydroxylase deficiency affects roughly one in 15,000 live births worldwide, with a carrier frequency near one in 60 in many populations. The birth prevalence is higher in communities with a founder effect — for example among Yupik Eskimos, Ashkenazi Jewish, Hispanic and Italian populations — and it is these pockets that skew local screening programmes. [6]
The condition is autosomal recessive, so the strongest risk factor is having affected parents or being born into a known carrier family. A previously affected child in the family is a flag for prenatal counselling and for measuring 17-hydroxyprogesterone on day-of-life bloods rather than waiting for the routine screen. Prematurity is not a cause, but it confounds the screen — 17-hydroxyprogesterone is physiologically high in sick and preterm infants, which is why birthweight- and gestation-adjusted cutoffs and second-tier testing exist. [5] [11]
VIRIL
Androgen excess in 21-OHD and 11β-OHD — clitoromegaly, rapid growth, advanced bone age
Salt-wasting: hyponatraemia, hyperkalaemia, hypoglycaemia, hypotension
Plasma renin is markedly elevated in the salt-wasting form — guides mineralocorticoid dosing
Parents carry an emergency intramuscular hydrocortisone injection for illness
Over-treatment with glucocorticoid suppresses height velocity — a key monitoring signal
Over a century of Swedish registry data show that early diagnosis through newborn screening has transformed outcomes, shifting death from unrecognised salt-wasting crises into the rare event and improving the lifetime profile of the disease. The trade-off of screening is the false-positive burden, which falls hardest on preterm and sick infants and is the reason programmes have moved to second-tier liquid chromatography-tandem mass spectrometry and, increasingly, molecular testing. [6] [11]
Pathophysiology
To understand why these babies salt-waste and virilise, picture the adrenal cortex as a single cholesterol pipeline that forks into cortisol, aldosterone, and androgens. In 21-hydroxylase deficiency the fork that leads to cortisol and aldosterone is blocked at one shared step, so the cholesterol that would have become cortisol and aldosterone is diverted down the androgen fork instead. [2]
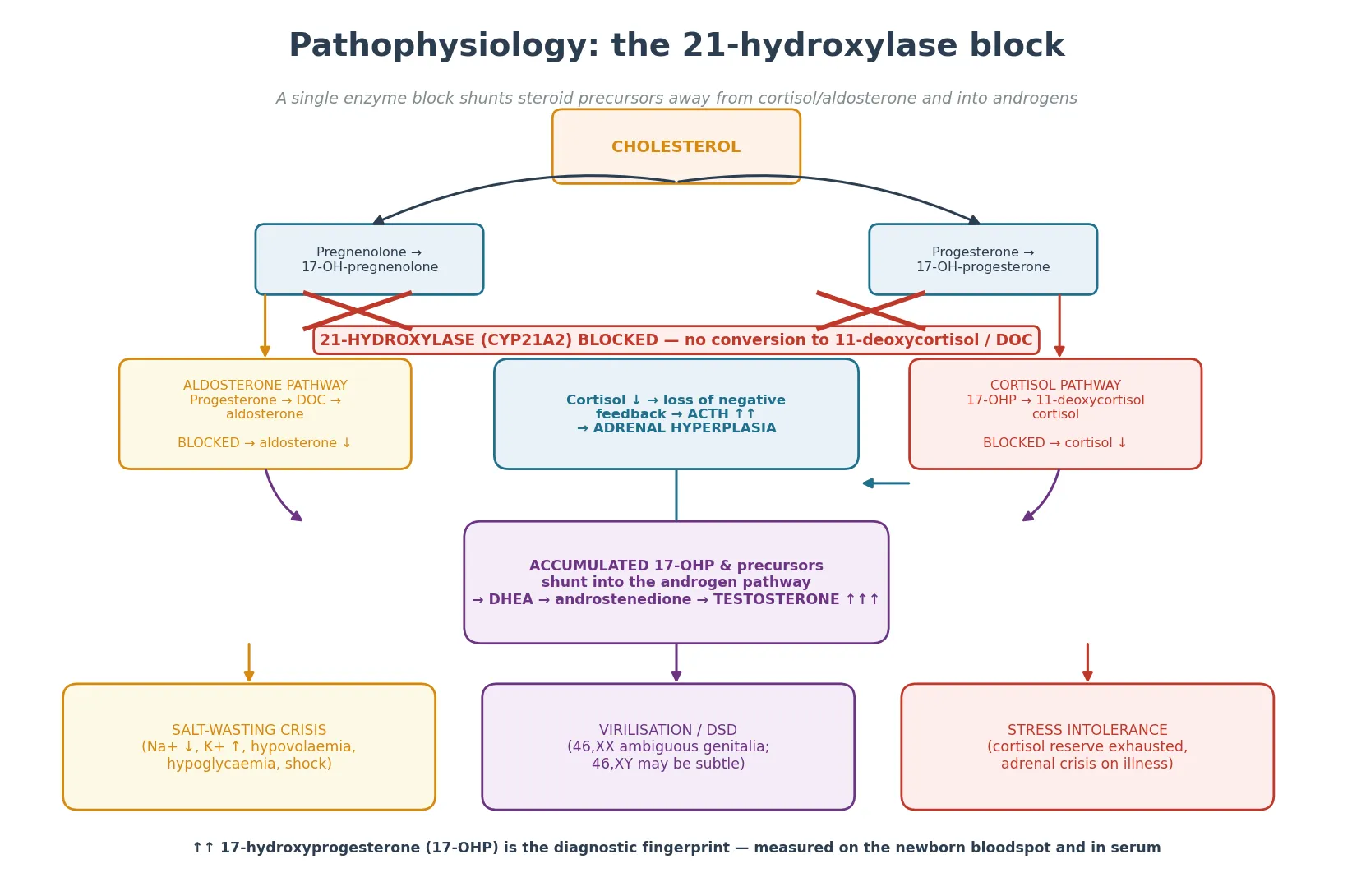
The cortisol fall removes negative feedback on the pituitary, so ACTH climbs and stimulates the now-hyperplastic cortex to churn out ever more precursor. The precursor that cannot cross the block is 17-hydroxyprogesterone, which is why a markedly elevated 17-hydroxyprogesterone is the biochemical fingerprint of 21-hydroxylase deficiency. Some of that precursor spills into androstenedione and testosterone, which virilise. [2] [3]
The aldosterone story depends on the severity of the block. In the salt-wasting form, the aldosterone pathway is also blocked, so aldosterone is low and the kidney cannot retain sodium or excrete potassium. The result is hyponatraemia, hyperkalaemia, metabolic acidosis, volume depletion and shock — usually between one and three weeks of life, once the maternal salt and glucocorticoid contribution has cleared. In simple-virilising disease, enough residual activity spares aldosterone, so the child virilises but does not salt-waste. [1]
Salt-wasting 21-OHD
- Both cortisol and aldosterone blocked
- Hyponatraemia with hyperkalaemia, hypoglycaemia, acidosis
- Presents day 7–21 in any sex; no genital clue in boys
- Volume-depleted, hypotensive, collapsing
Simple-virilising 21-OHD
- Cortisol low, aldosterone adequate
- Androgen excess virilises but no overt salt loss
- 46,XX genital ambiguity is the presenting clue
- Still adrenal-crisis prone under stress
Clinical Presentation
Most newborns with classic salt-wasting 21-hydroxylase deficiency look well for the first few days, because maternal cortisol and aldosterone cross the placenta. As that supply clears and the baby's own production fails, the picture darkens: poor feeding, vomiting, lethargy, then frank shock. The male neonate has no genital clue, so he is the one most often mislabelled as sepsis or pyloric stenosis while the adrenal crisis deepens. [1]
The salt-wasting crisis is biochemical as much as it is clinical. Sodium falls, potassium rises, glucose falls, and a metabolic acidosis develops as perfusion fails. Hypoglycaemia compounds the picture because cortisol normally supports gluconeogenesis. The blood pressure is low and the peripheral perfusion poor. Any term neonate presenting in shock with this electrolyte pattern has salt-wasting congenital adrenal hyperplasia until proven otherwise. [2]
The female neonate usually declares herself at birth by her ambiguous genitalia: clitoromegaly, posterior labial fusion, and a single urogenital sinus. Internal Müllerian structures (uterus, fallopian tubes, upper vagina) are present and normal because the gonads are ovaries and there is no anti-Müllerian hormone. The Prader stage grades the degree of virilisation from I (mild clitoromegaly) to V (complete masculinisation). This is the single most common cause of 46,XX disorder of sex development. [10]
Non-classic 21-hydroxylase deficiency declares itself later, outside the newborn period. The child presents with precocious pubarche, rapid linear growth with an advanced bone age, and — in adolescent girls — hirsutism, acne, oligomenorrhoea and a polycystic-ovary-syndrome-like picture. It is the diagnosis to reach for when the androgen excess is mild and the electrolytes are normal. [7]
Differential Diagnosis
The differential depends on which limb of the disease the child presents with. For the salt-wasting or shocked neonate, the universal mimic is sepsis, followed by pyloric stenosis, inborn errors of metabolism, congenital renal salt-wasting, and other causes of adrenal insufficiency. The discriminating features are the electrolyte pattern (hyponatraemia with hyperkalaemia points to aldosterone deficiency) and the glucose (cortisol deficiency drags glucose down). [1] [4]
For ambiguous genitalia in a 46,XX infant, the competing diagnoses are other virilising disorders — placental aromatase deficiency, maternal virilising tumours or androgen exposure — and these are usually distinguishable by a normal 17-hydroxyprogesterone. For a 46,XY infant with undervirilisation, the differential includes androgen insensitivity, 5α-reductase deficiency, gonadal dysgenesis and the rarer enzyme blocks such as 17α-hydroxylase and StAR defects that impair androgen synthesis. [10]
Points to CAH
- Hyponatraemia with hyperkalaemia in a sick neonate
- Hypoglycaemia alongside the electrolyte derangement
- 46,XX with clitoromegaly / posterior labial fusion
- Rapid growth and advanced bone age in an older child
- Family history of CAH or unexplained neonatal death
Points to a mimic
- Sepsis: risk factors, positive cultures, normal 17-OHP
- Pyloric stenosis: projectile vomiting, hypochloraemic alkalosis, palpable mass
- Pseudo-hypoaldosteronism: hyperkalaemia despite HIGH aldosterone
- Aromatase deficiency / maternal androgens: 46,XX virilisation with normal 17-OHP
- Androgen insensitivity: 46,XY undervirilisation, normal 17-OHP
A useful bedside discriminator is the relationship between sodium and potassium. Pyloric stenosis classically gives a hypokalaemic, hypochloraemic metabolic alkalosis — the opposite electrolyte picture to salt-wasting CAH. Renal salt-wasting and pseudo-hypoaldosteronism produce hyperkalaemia but aldosterone is high rather than low. Reaching for adrenal insufficiency at the same time as sepsis, rather than after it, is the habit that saves the neonate. [4]
Clinical & Bedside Assessment
The assessment of a suspected CAH neonate runs on two tracks at once: recognise the emergency and gather the data that will confirm it. Start with the ABCDE — airway, breathing, circulation — because the salt-wasting crisis is a volume-depleted, shock state. Capillary refill, heart rate, blood pressure, and conscious level tell you how sick the child is in front of you. [4]
The external genital examination is the second track and is done on every newborn. Inspect the phallus (size, chordee), the labioscrotal folds (fusion, rugosity, pigmentation), and the position of the urethral opening. Palpate for gonads in the labioscrotal folds — a palpable gonad almost always means testicular tissue, which rules out 46,XX CAH and redirects the work-up. A 46,XX baby with clitoromegaly and fused labia but no palpable gonads is virilising CAH until proven otherwise. [10]
Measure weight, length and head circumference and plot them, because both androgen excess (rapid growth, advanced bone age) and glucocorticoid over-replacement (growth suppression) declare themselves on the growth chart. Check the blood pressure in all four limbs — hypertension redirects the diagnosis toward 11β- or 17α-hydroxylase deficiency rather than 21-hydroxylase. Look for hyperpigmentation of the scrotum, labia and nipple line, which reflects chronic ACTH excess. [1]
Investigations
The bedside and laboratory tests come in tiers, because the newborn screen result, the serum 17-hydroxyprogesterone, and the electrolyte panel together make the diagnosis within hours. [1]
First-hour investigation bundle for suspected CAH
Capillary or venous gas — metabolic acidosis, hypoglycaemia
Electrolytes — hyponatraemia with hyperkalaemia is the fingerprint
Glucose — cortisol deficiency drags glucose down; treat it
Serum 17-hydroxyprogesterone — markedly elevated confirms 21-OHD
ACTH, plasma renin activity, aldosterone — confirms adrenal axis
Androgens (androstenedione, testosterone) — quantifies androgen excess
Karyotype / SNP array + pelvic ultrasound — confirms 46,XX with uterus
CYP21A2 molecular testing — defines genotype, informs family counselling
The newborn screen measures 17-hydroxyprogesterone on the dried bloodspot, collected on day 2 to 3 in most programmes. It is sensitive but imperfect: it misses some classic cases — particularly simple-virilising disease and infants with delayed salt-wasting — and it over-flags preterm, sick and stressed infants whose 17-hydroxyprogesterone is physiologically high. That is why modern programmes add a second-tier liquid chromatography-tandem mass spectrometry and, increasingly, CYP21A2 molecular testing to cut false positives and lift sensitivity. [5] [8] [11]
Cases of CAH missed by newborn screening (Minnesota)
JAMA
Population-based review of all CAH cases in Minnesota over 11 years, comparing those detected by newborn screening with those missed (Sarafoglou 2012).
Key finding
Newborn screening detected most, but not all, classic cases. Simple-virilising disease and infants who salt-wasted after the screen were over-represented among the missed cases.
Practice change
A passed newborn screen does not exclude CAH. Any neonate with vomiting, collapse, or a salt-wasting electrolyte pattern needs an urgent serum 17-hydroxyprogesterone regardless of the screen.
Serum 17-hydroxyprogesterone is the confirmatory test and is markedly elevated (often into the hundreds or thousands of nmol per litre) in classic 21-hydroxylase deficiency. Plasma renin is high and aldosterone low in the salt-wasting form. ACTH is elevated. A karyotype and a pelvic ultrasound confirm a 46,XX complement with a uterus, and CYP21A2 molecular testing defines the genotype, which correlates well with phenotype and underpins prenatal and sibling counselling. An ACTH stimulation test is reserved for borderline or non-classic cases where the basal 17-hydroxyprogesterone is equivocal. [9]
Management — Resuscitation
A salt-wasting crisis is a volume-depleted, cortisol-deficient shock state, and it kills if untreated. Resuscitation runs on three tracks at once: restore the circulating volume, replace the missing cortisol, and correct the electrolytes and glucose. [1] [4]
Restore volume with 10 to 20 mL per kg of isotonic saline repeated to restore perfusion, then start a maintenance fluid that contains sodium. Treat hypoglycaemia with intravenous dextrose. The single decisive drug is hydrocortisone intravenously: 25 mg stat in a neonate, then 50 to 100 mg per square metre per day by continuous infusion or divided doses. Hydrocortisone has enough mineralocorticoid activity at these stress doses to begin restoring sodium retention, so fludrocortisone is not needed in the acute phase. [4]

Hyperkalaemia in salt-wasting CAH usually corrects as volume, cortisol and aldosterone activity are restored; it is rarely dangerous enough to need specific hypokalaemic therapy, but monitor the ECG if the potassium is very high. Cover sepsis with cultures and empiric antibiotics, because sepsis and adrenal crisis are clinically indistinguishable in a sick neonate. Escalate to a paediatric intensive care setting if shock persists after the first fluid bolus and steroid dose. [4]
Across Endocrine Society (Speiser 2010; Bornstein 2016), ESPE and Australasian guidance, the acute salt-wasting crisis is treated with isotonic volume, parenteral hydrocortisone at stress doses, glucose correction, and sepsis cover. Local protocols differ on the exact hydrocortisone infusion regimen and on the threshold for intensive care, but the principle — volume, hydrocortisone, electrolyte and glucose correction in parallel — is universal. [1] [4]
Management — Definitive & Stepwise
Once the child is stable, definitive management is lifelong hormone replacement aimed at three goals: replace cortisol, replace aldosterone, and suppress the ACTH-driven androgen excess — all while avoiding the growth suppression and iatrogenic Cushing that come from over-treatment. The pathway runs in stages. [1]
Definitive pathway for confirmed CAH
Acute resuscitation with volume and parenteral hydrocortisone
Confirm with serum 17-OHP, ACTH, renin, androgens, karyotype and CYP21A2 testing
Start oral hydrocortisone 10–15 mg/m²/day in three divided doses
Add fludrocortisone 0.05–0.2 mg/day; neonates also need Na+ 2–3 mmol/kg/day
Refer to a multidisciplinary DSD team for counselling and genitoplasty decision
Build a stress-dose and emergency-injection plan; MedicAlert and school care plan
Long-term monitoring of growth, bone age, electrolytes, renin, 17-OHP and androgens
Structured transition to adult endocrinology in late adolescence
Glucocorticoid replacement is the cornerstone and uses hydrocortisone 10 to 15 mg per square metre per day in three divided doses in growing children, because hydrocortisone is short-acting and lets you titrate without the growth-suppressive burden of long-acting steroids. The dose is split to mimic the cortisol rhythm, with the largest dose in the morning. Longer-acting glucocorticoids such as prednisolone or dexamethasone are reserved for older patients or for specific suppression needs, because they suppress growth more readily. [1]
Mineralocorticoid replacement uses fludrocortisone 0.05 to 0.2 mg once daily, titrated to the plasma renin. Neonates and infants also need oral sodium supplementation at 2 to 3 mmol per kg per day, because their milk intake is sodium-poor and they cannot conserve sodium on fludrocortisone alone until the diet matures. As solid food introduces sodium, the supplement is weaned. [1] [4]
Hydrocortisone
The aim of chronic dosing is a tightrope. Too little glucocorticoid leaves ACTH high, androgens unchecked, and the child overgrowing with an advanced bone age that steals final height. Too much glucocorticoid suppresses growth and produces iatrogenic Cushing. The monitoring tools are the growth chart, the bone age, and the biochemistry — renin for mineralocorticoid adequacy, and 17-hydroxyprogesterone and androstenedione for glucocorticoid adequacy — interpreted together rather than chased to a single number. [1] [3]
Specific Subtypes & Scenarios
The highest-yield scenarios for exams and for the bedside are the four below, because they are the ones that present, collapse, and are misdiagnosed. [1]
Classic salt-wasting 21-hydroxylase deficiency is the archetype. The 46,XX infant is flagged by clitoromegaly and labial fusion at birth, but the 46,XY infant is genitally normal and presents at one to three weeks with vomiting, collapse, and the classic electrolyte pattern. The first dose of hydrocortisone and the first fluid bolus are the most important interventions of the child's life. [1]
Classic simple-virilising 21-hydroxylase deficiency spares aldosterone enough to avoid overt salt-wasting, so it may surface only through the genital ambiguity of a 46,XX infant or, later, through androgen excess. These children still need glucocorticoid replacement and a stress-dose plan, because their cortisol reserve is insufficient under illness. [9]
Non-classic 21-hydroxylase deficiency declares itself outside the newborn period with precocious pubarche, rapid growth and an advanced bone age in children, and with hirsutism, acne and oligomenorrhoea in adolescents. A polycystic-ovary-syndrome look-alike with a family history of CAH or an early-morning 17-hydroxyprogesterone above the equivocal range is the clue, confirmed by an ACTH stimulation test. [7]
11β-hydroxylase deficiency is the hypertension variant: 11-deoxycorticosterone accumulates and acts as a mineralocorticoid, so the child is hypertensive and hypokalaemic rather than salt-wasting, while still virilised. Reaching for it depends on measuring the blood pressure in a virilised infant and noticing that the electrolytes do not fit the salt-wasting pattern. [2]
Complications & Pitfalls
The complications divide into those of the disease and those of its treatment. Untreated or under-treated CAH brings adrenal crisis, short adult stature from uncontrolled androgen excess and premature epiphyseal fusion, infertility and menstrual disturbance from ongoing androgen excess, and the psychosocial burden of a disorder of sex development. Over-treated CAH brings iatrogenic Cushing, growth suppression, osteopenia and metabolic disturbance from chronic glucocorticoid excess. [3]
Disease pitfalls
- Missing salt-wasting CAH in a genitally normal boy labelled 'sepsis'
- Treating pyloric stenosis when the electrolytes are hyponatraemic/hyperkalaemic
- Forgetting that a passed newborn screen does not exclude CAH
- Letting androgen excess run and stealing final height
Treatment pitfalls
- Over-replacing glucocorticoid and suppressing linear growth
- Forgetting fludrocortisone and sodium supplement in the neonate
- Failing to give a stress dose during illness or before surgery
- Chasing a single 17-OHP number instead of the whole child
The most lethal pitfall is the missed male neonate. Because he has no genital clue, he is treated as sepsis or pyloric stenosis while his adrenal cortex fails, and he arrests in salt-losing shock. The countermeasure is a low threshold to send a 17-hydroxyprogesterone in any neonate with unexplained vomiting, poor weight gain, or collapse — and to give a dose of hydrocortisone empirically in a sick, hyponatraemic child while the result is pending. [8]
The second pitfall is the sick child on treatment. A child with CAH cannot mount a cortisol response to the stress of a fever, a gastroenteritis, or an operation. They need a stress dose: two to three times their usual hydrocortisone for illness, and parenteral hydrocortisone if they are vomiting or cannot absorb oral medication. Parents must carry an emergency intramuscular hydrocortisone injection and know how to use it. [4]
Prognosis & Disposition
With early diagnosis and disciplined replacement, the outlook for congenital adrenal hyperplasia is good. Children with 21-hydroxylase deficiency can expect normal intellectual development, near-normal final height if the glucocorticoid dose is well balanced, and the possibility of fertility — particularly in males and in females whose androgen excess is well controlled. The single biggest modifiable factor is whether the diagnosis is made before a salt-wasting crisis. [1] [6]
Final height remains the long-term battleground. Both under-treatment (androgen-driven skeletal maturation) and over-treatment (glucocorticoid-driven growth suppression) steal centimetres, so the dose is a daily negotiation between two opposing harms. Fertility in females is affected by the degree of androgen control and by any genital surgery, which is why modern practice leans toward shared, deferred decisions about genitoplasty. [3] [10]
Disposition is lifelong. The child leaves the acute admission on hydrocortisone, fludrocortisone and (in infancy) sodium, with a stress-dose plan, an emergency injection, a MedicAlert identifier, a school care plan, and a named paediatric endocrinologist. Structured transition to adult endocrinology in late adolescence carries the dosing, the monitoring and the counselling forward, and is now a measured quality marker of CAH care. [1]
Special Populations
The affected sibling and the fetus at risk are the planned pathway. A family with one affected child is offered prenatal or pre-implantation genetic diagnosis and, in some programmes, antenatal dexamethasone to the mother to suppress fetal androgen production and prevent virilisation of an affected 46,XX fetus. Antenatal dexamethasone is contentious — it is given before the fetal sex and genotype are known, and it carries maternal and fetal risks — so modern counselling weighs it carefully against the option of early postnatal treatment and shared genitoplasty decisions. [1] [10]
The preterm or sick neonate confounds the screen. 17-hydroxyprogesterone is physiologically elevated in prematurity and illness, which inflates false positives and can mask a true case if a single-tier cutoff is used. Birthweight- and gestation-adjusted cutoffs and second-tier liquid chromatography-tandem mass spectrometry reduce the noise, and molecular testing is increasingly added when the first two tiers disagree. [5] [11]
The adolescent and young adult carries the cumulative burden of a chronic disease into transition. For girls this includes the legacy of genital surgery, questions of sexual function and identity, and the management of androgen excess against glucocorticoid burden; for both sexes it includes fertility counselling, adherence to a twice-or-thrice-daily medication, and the need for an emergency plan that travels with them. A trauma-informed, honest approach to the disorder-of-sex-development label is part of good transition care. [10]
Indigenous, migrant and remote populations face both a higher founder-effect prevalence in some communities and the logistics of accessing a specialist service. Newborn screening is the great equaliser here, but it only works if the result is acted on, which depends on follow-up, transport, and culturally safe counselling. Equity of access to endocrine care and to a stress-dose plan is a determinant of survival in remote settings. [5]
Evidence, Guidelines & Regional Differences
The evidence base for congenital adrenal hyperplasia rests on a long registry tradition and on the Endocrine Society guidelines. The Endocrine Society 2010 guideline on 21-hydroxylase deficiency set the modern standard for diagnosis, hormone replacement, stress dosing and the surgical and psychosocial approach, and the 2016 primary adrenal insufficiency guideline extended the adrenal-crisis principles that govern the salt-wasting presentation. [1] [4]
Newborn screening transformed the disease. The Swedish 26-year prospective population-based study showed that universal 17-hydroxyprogesterone screening detected the great majority of classic cases and reduced the death and morbidity of unrecognised salt-wasting, while the century-long Swedish cohort placed the modern era against a historical baseline in which CAH was a frequent cause of neonatal death. [5] [6]
The Minnesota review tempered the optimism. It showed that newborn screening misses a real minority of cases — over-represented by simple-virilising disease and by infants whose salt-wasting declared after the screen — which is why a passed screen never lowers the threshold to investigate a vomiting or collapsing neonate. [8] Second-tier testing with liquid chromatography-tandem mass spectrometry, and increasingly CYP21A2 molecular testing, now cut the false-positive burden that screening places on preterm and sick infants. [11]
Australia and Aotearoa New Zealand screen all newborns for 21-hydroxylase deficiency on the day-2 to -3 bloodspot, with birthweight-adjusted cutoffs for preterm infants and second-tier testing where indicated. Acute salt-wasting crises are managed in regional and tertiary paediatric services with volume, parenteral hydrocortisone at stress doses, and sepsis cover, with retrieval to a paediatric endocrine centre. Replacement follows the Endocrine Society framework: hydrocortisone 10 to 15 mg per square metre per day, fludrocortisone, and sodium supplement in infancy. [1] [5]
The remaining controversies are practical and ethical: the exact glucocorticoid preparation and regimen that best balances androgen control against growth; the role of new therapies such as modified-release hydrocortisone and adrenal-blocking agents; whether and when to offer antenatal dexamethasone; and the timing, necessity and consent model for feminising genitoplasty. These are genuinely contested, and a fellowship candidate names the debate rather than pretending it is settled. [3] [10]
Exam Pearls
One-sentence answer: the approach to suspected CAH
A neonate with ambiguous genitalia or a salt-wasting collapse at one to three weeks has congenital adrenal hyperplasia until proven otherwise: send an urgent 17-hydroxyprogesterone, resuscitate with volume and parenteral hydrocortisone, confirm with electrolytes, renin, ACTH, karyotype and CYP21A2 testing, then replace hydrocortisone, fludrocortisone and sodium with a stress-dose and emergency-injection plan.
Epidemiology and screening
- Classic CAH birth prevalence ~1 in 15,000; ~95% is 21-hydroxylase deficiency
- ~75% of classic 21-OHD is salt-wasting
- Newborn screen = 17-OHP on the day-2/3 bloodspot; second-tier LC-MS/MS for preterm/sick
- A passed screen does NOT exclude CAH (Sarafoglou 2012)
The crisis
- Hyponatraemia + hyperkalaemia + hypoglycaemia + shock at 1–3 weeks = salt-wasting CAH
- 46,XY has NO genital clue — the missed-male is the lethal trap
- Treat empirically: volume 10–20 mL/kg saline + hydrocortisone 25 mg IV stat
Doses to know
- Acute hydrocortisone: 25 mg IV stat (neonate), then 50–100 mg/m²/day
- Chronic hydrocortisone: 10–15 mg/m²/day in 3 divided doses
- Fludrocortisone: 0.05–0.2 mg/day; neonate Na+ 2–3 mmol/kg/day
- Stress: 2–3× hydrocortisone for illness; parenteral if vomiting
Frequently misremembered facts, stated correctly: pyloric stenosis gives a hypokalaemic hypochloraemic alkalosis, the opposite of salt-wasting CAH. A passed newborn screen does not exclude CAH. Hydrocortisone at stress doses provides enough mineralocorticoid activity that fludrocortisone is not needed in the acute crisis. And the 46,XY neonate has no genital clue — the diagnosis is missed until he salt-wastes. [8]
The lesion-to-sign pairings are the fastest route to a bedside answer and the highest-yield material for a written or oral question: salt-wasting with hyperkalaemia is 21-hydroxylase; hypertension with virilisation is 11β-hydroxylase; hypertension with undervirilisation is 17α-hydroxylase; and a 46,XX virilised infant with a uterus and no palpable gonad is 21-hydroxylase deficiency until proven otherwise. [2] [9]
References
- [1]Speiser PW; Azziz R; Baskin LS; et al Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab, 2010.PMID 20823466
- [2]White PC; Speiser PW Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev, 2000.PMID 10857554
- [3]Merke DP; Bornstein SR Congenital adrenal hyperplasia. Lancet, 2005.PMID 15964450
- [4]Bornstein SR; Allolio B; Arlt W; et al Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2016.PMID 26760044
- [5]Gidlöf S; Wedell A; Guthenberg C; et al Nationwide neonatal screening for congenital adrenal hyperplasia in Sweden: a 26-year longitudinal prospective population-based study. JAMA Pediatr, 2014.PMID 24733564
- [6]Gidlöf S; Falhammar H; Thilén A; et al One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet Diabetes Endocrinol, 2013.PMID 24622265
- [7]Witchel SF; Azziz R Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol, 2010.PMID 20671993
- [8]Sarafoglou K; Banks K; Kyllo C; et al Cases of congenital adrenal hyperplasia missed by newborn screening in Minnesota. JAMA, 2012.PMID 22692165
- [9]New MI; Abraham M; Gonzalez B; et al Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A, 2013.PMID 23359698
- [10]Houk CP; Hughes IA; Ahmed SF; et al Summary of consensus statement on intersex disorders and their management. Pediatrics, 2006.PMID 16882833
- [11]Engström K; Zetterström RH; Wedell A; et al Neonatal Screening for CAH in Sweden—Results of Implementing Second-Tier Testing. Int J Neonatal Screen, 2026.PMID 42201221