Paeds · endocrinology-diabetes-and-growth
Constitutional delay and familial short stature
Also known as Constitutional delay of growth and puberty (CDGP) · Familial short stature · Self-limited delayed puberty · Constitutional growth delay · Normal-variant short stature
A fellowship approach to the two commonest normal variants of short stature: recognise familial short stature as a low polygenic height set-point with a normal bone age and constitutional delay of growth and puberty as a slowed hypothalamic-pituitary-gonadal clock with a delayed bone age, separate both from the pathological causes by trajectory and a focused workup, and manage almost all children with reassurance and surveillance while reserving short-course testosterone or oxandrolone for the distressed adolescent and growth hormone only for true deficiency or idiopathic short stature.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A seven-year-old boy is brought to clinic because he is the shortest in his class. Both parents are short, his father began shaving at seventeen, and his growth chart shows him tracking just below the third centile since toddlerhood with a normal growth velocity. Down the corridor, a fourteen-year-old boy with no signs of puberty is distressed that he is still a child among his peers; his father went through puberty late, his bone age reads eleven years, and his height is appropriate for that bone age. In both rooms the task is the same: confirm that this is a normal variant of growth, exclude the pathological mimics, and decide whether anything needs to be done beyond reassurance and surveillance. The skill is not to over-investigate normal biology, nor to miss the child whose short stature or delayed puberty is the first sign of disease. [1] [9]
B · O · N · E · S for the short child
Overview & Definition
Short stature is defined as a height more than two standard deviations below the mean for age and sex, which places the child below the third centile on a population growth chart. It is a presenting complaint rather than a diagnosis, and the clinician's first task is to decide whether the shortness is a normal variant of growth or the sign of an underlying disorder. The two commonest normal variants are familial short stature and constitutional delay of growth and puberty, and together they account for a large proportion of the short children seen in general paediatric and endocrine practice. Recognising them is a core skill because the appropriate management is reassurance, and the principal error is either over-investigating a normal child or, conversely, labelling a pathological process as constitutional. [1] [4]
Familial short stature is the straightforward expression of a child's genetic height potential. Height is one of the most heritable of human traits — roughly eighty per cent of the variation in adult height between individuals is attributable to inherited polygenic factors, with hundreds of common gene variants each contributing a small effect. A child of short parents has inherited a low polygenic set-point, grows normally at a normal velocity, and ends up short in a way that is entirely appropriate for the family. The bone age matches the chronological age, puberty begins on schedule, and the final adult height falls within the calculated mid-parental (target) height range. There is no disease, no deficiency, and no distortion of growth — only a normal child at the short end of the normal range. [2] [4]
Constitutional delay of growth and puberty is a variant of tempo rather than of set-point. The child has a slowed biological clock: the hypothalamic-pituitary-gonadal axis, dormant since infancy, reactivates late, so puberty begins later than in peers and the pubertal growth spurt is delayed. The bone age lags behind the chronological age and typically matches the height age, which is why the child is short for chronological age but on track for bone age. The characteristic feature is a late but prolonged growth spurt that carries the child to a final adult height within the normal range and, crucially, within the family target range. Constitutional delay is common in boys — who present far more often than girls, largely because the psychosocial impact of delayed puberty is more pronounced in adolescent males — and there is usually a family history of late puberty in a parent or first-degree relative. [9] [11]
Classification
Short stature is best classified by two clinical axes — whether the child is proportionate, and whether the growth velocity is normal — because these two questions rapidly sort the normal variants from the pathological causes. The figure lays out the classification: a short child is first assessed for disproportion (pointing to a skeletal dysplasia), then for growth velocity. A short child with a normal growth velocity and a proportionate build falls into the normal-variant group, where familial short stature and constitutional delay sit, while a child with an abnormal velocity or a downward drift across centiles falls into the pathological group, which spans primary endocrine causes, systemic and secondary causes, and the genetic syndromes. [1] [5]
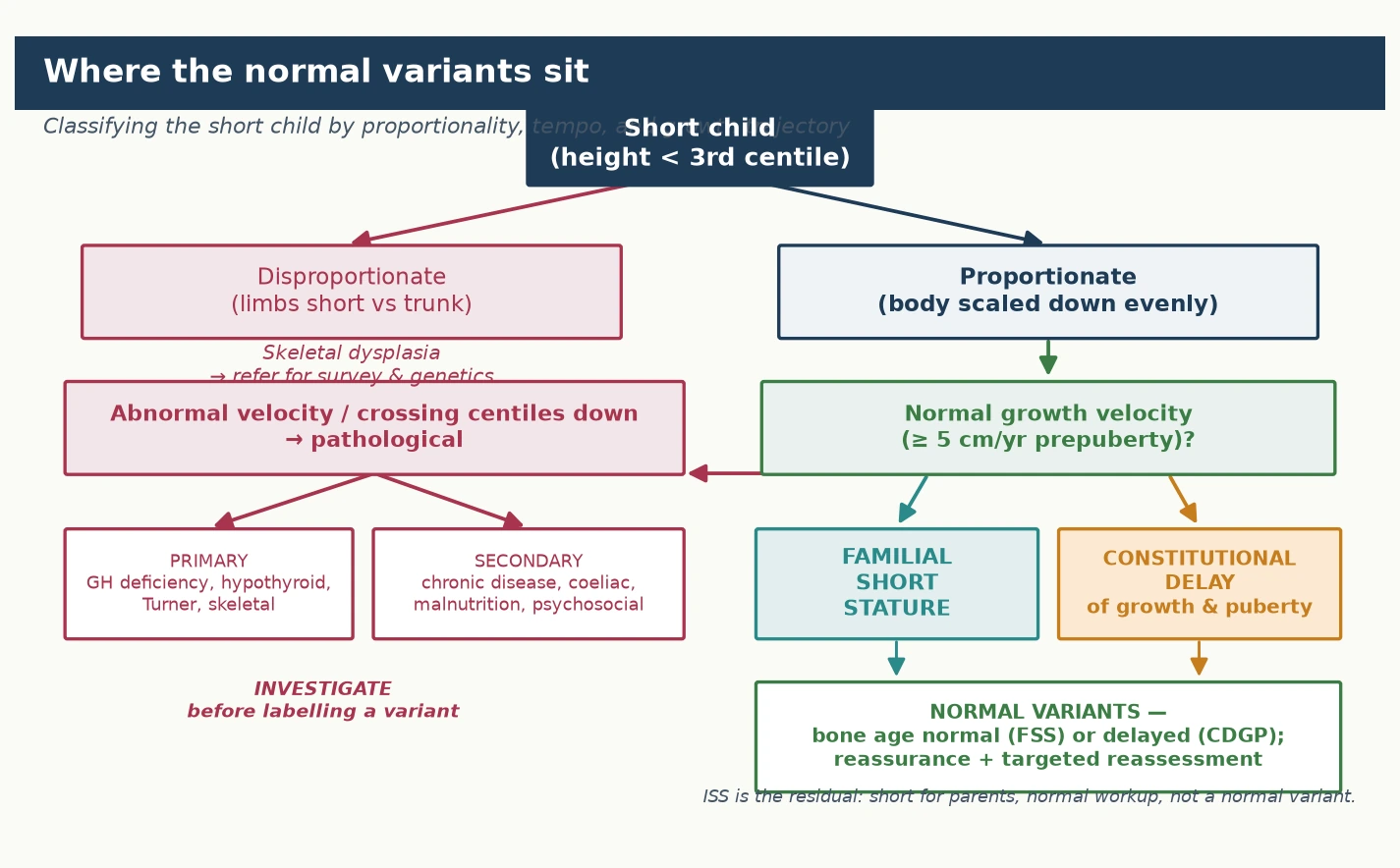
The two normal variants are themselves distinguished by the bone age and the growth trajectory, and holding the distinction firmly prevents both over-treatment and missed diagnosis. In familial short stature the bone age is normal and the child tracks parallel to the centile lines from early childhood; in constitutional delay the bone age is delayed and the child drifts down through the centiles in late childhood before the late growth spurt. Both preserve a normal growth velocity, and both reach a final adult height appropriate for the family — the difference is the timing of puberty and the bone-age maturation, not the height potential. The third category, idiopathic short stature, is a residual diagnosis for the child who is short for their parents and for the population, has had pathology excluded, and is not a normal variant; it is discussed in detail on the growth-hormone-deficiency page but is named here so that it is not confused with the two normal variants. [2] [10]
Familial short stature versus constitutional delay — the discriminator
- Familial short stature: short parents; short from early childhood; tracks parallel to the centiles; bone age equals chronological age; puberty on time; final height short but within the mid-parental target range.
- Constitutional delay of growth and puberty: often a family history of late puberty; normal birth weight then drifts down in childhood; bone age delayed (matches height age); puberty late; late but prolonged growth spurt; final height within the normal range and the family target.
- The discriminator: the bone age and the timing of puberty. Normal bone age and on-time puberty point to familial short stature; delayed bone age and late puberty point to constitutional delay. Both share a normal growth velocity and a normal final height for the family. [4] [9]
Epidemiology & Risk Factors
Short stature is one of the commonest reasons for referral to paediatric services, and the normal variants make up a substantial fraction of those referrals. Constitutional delay of growth and puberty is estimated to account for a significant proportion of boys referred for evaluation of short stature or delayed puberty, and it is far more frequently identified in boys than in girls — a sex imbalance thought to reflect both biology and the greater psychosocial visibility of delayed puberty in adolescent males. Familial short stature is similarly common wherever short parents have children, and the two conditions frequently overlap, because a child can inherit both a low height set-point and a slow tempo from their parents. [1] [5]
The dominant risk factor for both conditions is family history, and taking an accurate parental and pubertal history is the single highest-yield epidemiological step. For familial short stature, both parents are typically short, and measuring and plotting the parents' heights is essential — a child whose height is consistent with the mid-parental target range almost certainly has a normal variant rather than disease. For constitutional delay, a history that one or both parents, or a sibling, had a late growth spurt or was a "late bloomer" is strongly supportive; paternal history of late shaving or a late voice break, and maternal history of late menarche, are the useful markers. The inheritance of constitutional delay is complex and polygenic, with strong familial clustering but no single causative gene, which is why the family history rather than a genetic test makes the diagnosis. [9] [11]
There is an important overlap pattern that examiners test: a child can have both familial short stature and constitutional delay simultaneously, and this combination produces the child who is short for their family and whose puberty is late, whose final height lands at the lower end of the (already short) parental range. Recognising that the two variants are independent and can co-exist prevents the clinician from being falsely reassured by a normal bone age (which would suggest FSS alone) when a delayed puberty is also present, or from over-treating a child who simply carries both normal traits. [4] [5]
Pathophysiology
The two normal variants arise from different biological mechanisms, and understanding them clarifies why each behaves the way it does. The figure traces the two cascades side by side: familial short stature is a polygenic height set-point with normal growth-plate function, while constitutional delay is a slowed hypothalamic-pituitary-gonadal tempo with a bone age that lags behind chronology. [4] [8]
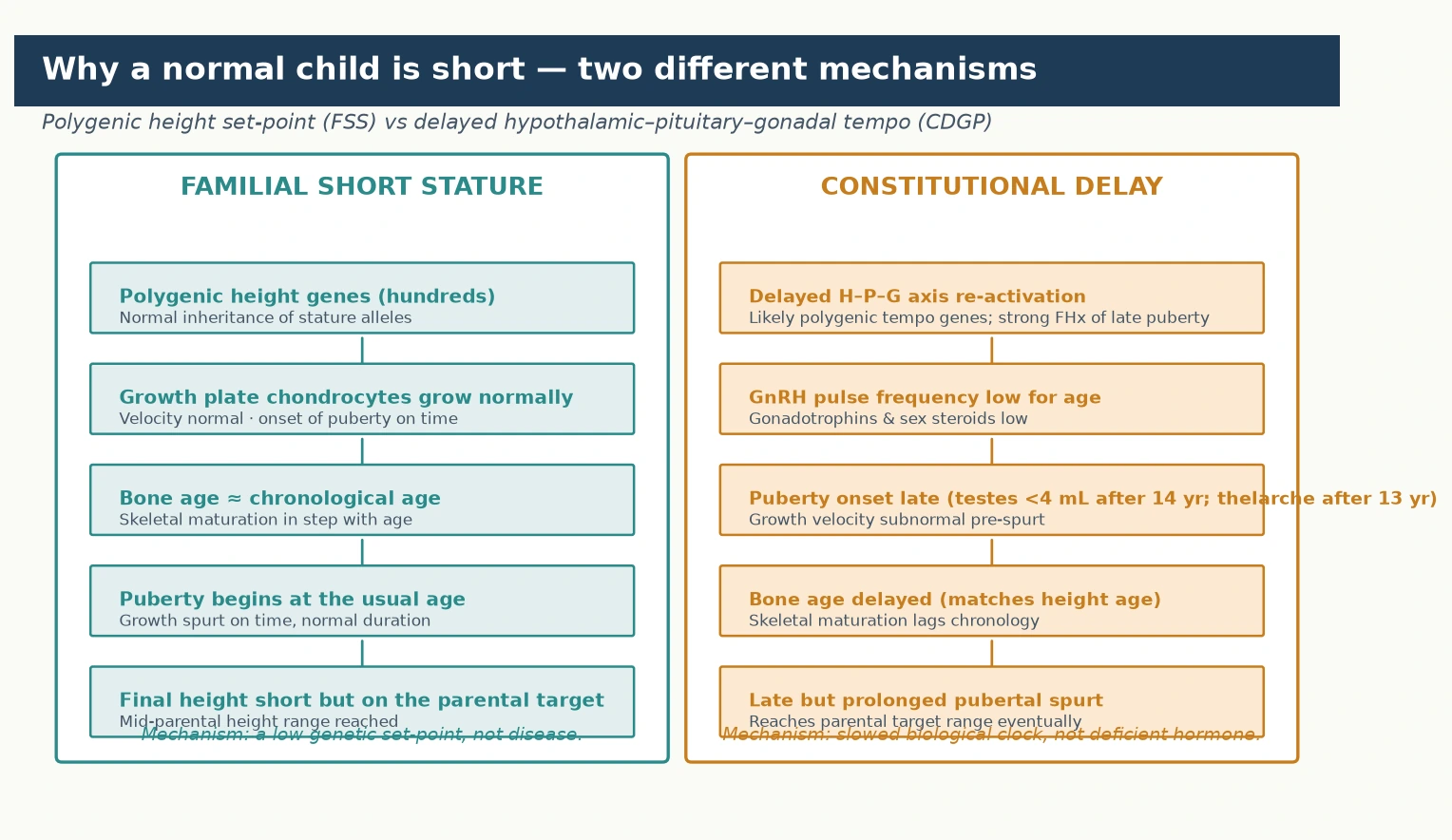
Familial short stature is the expression of inherited polygenic variation. Adult height is among the most heritable human traits, with genome-wide association studies identifying thousands of common variants — each of tiny individual effect — that collectively account for the majority of height variation between people. A child of short parents inherits a complement of these variants that sets a lower growth trajectory, but the growth plate functions entirely normally: chondrocytes proliferate and hypertrophy on schedule, endochondral ossification proceeds at a normal pace, and the bone age advances in lockstep with the chronological age. Puberty begins at the expected time, the pubertal growth spurt is of normal timing and magnitude, and growth ceases at the usual age when the epiphyses fuse. The child is short because the set-point is short, not because any part of the growth machinery is defective — which is exactly why no therapy aimed at the growth plate (growth hormone, for instance) meaningfully raises the final height of a true familial short stature child. [2] [4]
Constitutional delay is a disorder of timing, driven by a slowed reactivation of the hypothalamic-pituitary-gonadal axis. During childhood this axis is physiologically suppressed after the mini-puberty of infancy, and its reactivation — the onset of the pulsatile gonadotropin-releasing-hormone signalling that triggers puberty — is normally tightly timed. In constitutional delay this reactivation is shifted later, so the gonadotrophins (luteinising and follicle-stimulating hormone) and the sex steroids (testosterone in boys, oestrogen in girls) rise later than in peers. Because sex steroids drive the pubertal growth spurt and the maturation of the skeleton, their late rise means both the spurt and the bone-age advance are delayed. The bone age therefore lags behind the chronological age and typically matches the height age — the age at which the child's height would be average — which is the hallmark that distinguishes constitutional delay from familial short stature. Crucially, the growth plate itself is normal, the growth velocity before the spurt is preserved, and once puberty begins the spurt is late but prolonged, ultimately delivering the child to a final adult height within the normal range. [8] [9]
Clinical Presentation
The presentation of the two normal variants differs, and recognising each pattern at the bedside directs the workup and the reassurance. Familial short stature typically presents in the toddler or preschool years, when a parent or clinician notices that the child is shorter than peers. The birth weight and length were usually normal, the child settles onto a lower centile in the first two years of life, and from then on tracks parallel to the centile lines with a normal growth velocity. There are no symptoms of systemic disease, the appetite and energy are normal, the general examination is unremarkable, and puberty begins at the expected age. The parents are short, and the child's height sits comfortably within the mid-parental target range. The combination of short parents, a child tracking parallel from early childhood, and a normal growth velocity is the classic presentation, and it is largely a story told by the growth chart. [1] [4]
Constitutional delay presents later and more dramatically, usually in early adolescence, when the child — most often a boy — and the family become aware that puberty has not started while peers are well into it. The history often reveals a normal birth weight, a drift downward across the centiles in mid-childhood as peers begin their pre-pubertal growth acceleration, and then a child who is short and prepubertal at an age when classmates are growing rapidly. The family history frequently identifies a parent or sibling who was a late bloomer, with late menarche in the mother or late shaving and a late voice break in the father. On examination there are no signs of puberty — in a boy the testes remain below four millilitres in volume, and in a girl there is no breast budding — but the child is otherwise well, with a normal general examination and a height that is appropriate for the bone age. [9] [11]
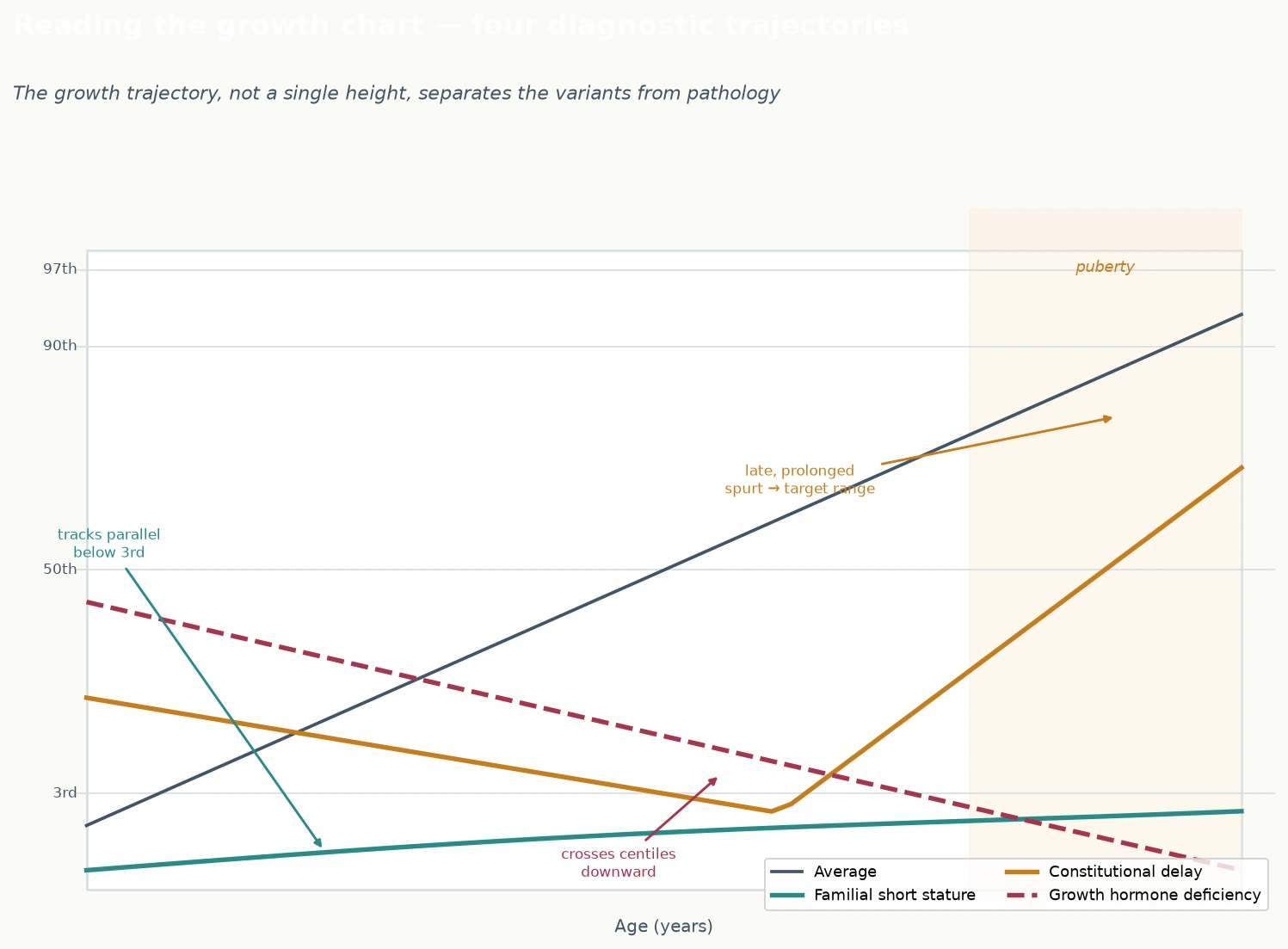
The growth chart is the single most informative clinical tool, and reading the trajectory correctly is the skill that separates the variants from the pathology. A normal-variant child grows at a normal velocity — generally at least five centimetres a year before puberty — and either tracks parallel to the centiles (familial short stature) or drifts down in childhood before the late spurt (constitutional delay). The cardinal red flag that excludes a normal variant is crossing centile lines downward: a child whose height is falling away from the centiles they previously tracked along is NOT a normal variant, however reassuring the family history, and demands investigation for growth hormone deficiency, hypothyroidism, chronic disease, coeliac disease, or psychosocial deprivation. The figure shows the four diagnostic trajectories — the normal variants and the pathological pattern — and learning to read them is the foundation of the assessment. [1] [5]
Differential Diagnosis
The differential diagnosis of the short child is broad, and the role of the clinician assessing a suspected normal variant is to exclude the pathological mimics efficiently and confidently. The highest-yield distinction is the growth velocity: a child growing at a normal rate along an expected trajectory is almost always a normal variant, while a child crossing centiles downward has a pathological cause until proven otherwise. Among the pathological causes, growth hormone deficiency is the principal endocrine mimic — it presents with a slow growth velocity, a delayed bone age, and a child who looks younger than their age, closely resembling constitutional delay. The difference is that the growth velocity is subnormal, and provocative growth hormone stimulation testing, together with insulin-like growth factor one levels, distinguishes the two. [2] [7]
Hypothyroidism, whether congenital or acquired, is a critical mimic because it is common, treatable, and easily missed. Acquired hypothyroidism can present with a slowing of growth velocity, a delayed bone age, and sluggishness or constipation, and a thyroid function test is part of the routine panel for any short child whose diagnosis is not immediately obvious. Turner syndrome must be on the differential for any short girl, even in the absence of the classic stigmata of webbed neck, broad chest, and lymphoedema, because short stature is often the only presenting feature. A karyotype or chromosomal microarray should be considered in a short girl with a delayed bone age before the short stature is labelled constitutional. Chronic disease — coeliac disease, inflammatory bowel disease, chronic kidney disease, and poorly controlled asthma — can present with short stature as the first sign, and a screening history for gastrointestinal symptoms, fatigue, and exercise tolerance, together with a coeliac screen and renal function, excludes the common offenders. [1] [5]
The differential of delayed puberty itself deserves attention, because constitutional delay and permanent hypogonadotrophic hypogonadism can be indistinguishable at presentation. Permanent hypogonadotrophic hypogonadism — including Kallmann syndrome with its associated hyposmia or anosmia and midline defects — presents with absent or stalled puberty, low gonadotrophins, and no underlying structural cause, and at the first presentation it may look exactly like constitutional delay. The discriminating features are a family history of late puberty (favouring constitutional delay), the presence of hyposmia or midline defects (favouring Kallmann), and ultimately the tempo of progression: constitutional delay enters and progresses through puberty spontaneously, whereas permanent hypogonadotrophic hypogonadism does not. Where doubt persists, watching the tempo with regular review, or undertaking provocative testing with gonadotropin-releasing hormone or human chorionic gonadotropin, resolves the question, and an over-hasty label of either should be avoided. [9] [10]
Clinical & Bedside Assessment
The assessment of a short child is built around three questions that the history, examination, and growth chart answer together. First, is the child growing at a normal velocity along an expected trajectory — because this is the gateway to a normal-variant diagnosis. Second, are the parents short, and is there a family history of late puberty — because the family history makes or breaks the diagnosis of the two variants. Third, are there any symptoms, signs, or past-history features that point to a pathological cause — because excluding the mimics is the essential safety net beneath the reassurance. A focused consultation answers all three and directs the bone age and blood tests that complete the workup. [1] [4]
The history captures the growth story, the family story, and the systemic story. Take the birth history — birth weight, gestation, and any perinatal complications — because intrauterine growth restriction or prematurity can shape the trajectory. Ask when the short stature was first noticed and obtain all previous growth measurements, because plotting serial heights to calculate the growth velocity is the single most important step. Measure and record the parents' heights and calculate the mid-parental (target) height, and ask specifically about the timing of puberty in the parents and siblings — late menarche in the mother, late shaving or a late growth spurt in the father. Then screen for systemic disease: appetite, energy, abdominal pain, diarrhoea or constipation, exercise tolerance, headaches or visual symptoms (raising pituitary or intracranial concerns), and the psychosocial environment. A thorough history alone frequently points to the diagnosis before any test is ordered. [1] [9]
The examination confirms proportionality and excludes the stigmata of disease. Measure the height accurately with a stadiometer, the weight, and the head circumference, and calculate the body mass index. Assess for disproportion by measuring the upper-to-lower body segment ratio and the arm span against height — a normal-variant child is proportionate, and disproportion points to a skeletal dysplasia. Examine for the dysmorphic features of Turner syndrome in girls (short stature, broad chest, widely spaced nipples, webbed neck, low posterior hairline), the stigmata of hypothyroidism (goitre, bradycardia, delayed reflexes, dry skin), and the signs of chronic disease. Perform a pubertal assessment using the Tanner staging, measuring testicular volume in boys with a Prader orchidometer and staging breast development in girls, because the absence of pubertal signs at the expected age is central to the diagnosis of constitutional delay. [4] [5]
Investigations
The investigation strategy for a suspected normal variant is deliberately focused, because the goal is to exclude the pathological mimics efficiently without over-investigating a normal child. The two cornerstone investigations are the growth chart — with serial measurements to confirm a normal velocity — and the bone age, read from a radiograph of the left hand and wrist. The bone age, assessed against standards such as the Greulich and Pyle atlas, is the discriminator between familial short stature (bone age equals chronological age) and constitutional delay (bone age delayed, matching the height age). A bone age within the normal range for chronological age in a child with short parents supports familial short stature, while a delayed bone age with an otherwise normal evaluation supports constitutional delay. [1] [8]
A targeted panel of blood tests excludes the common pathological mimics and is reasonable in any child whose diagnosis is not immediately clear from the growth chart and bone age. The panel includes a full blood count, a coeliac screen (tissue transglutaminase antibodies), renal and liver function, a thyroid function test, and an insulin-like growth factor one level as a screening test for growth hormone deficiency. In a short girl with a delayed bone age, a karyotype or chromosomal microarray should be considered to exclude Turner syndrome. These tests are directed at the common and treatable causes of short stature — coeliac disease, hypothyroidism, growth hormone deficiency, Turner syndrome, and chronic kidney or liver disease — and a normal panel, combined with a normal growth velocity and a supportive family history, secures the diagnosis of a normal variant. [2] [5]
Provocative testing and imaging are reserved for the child in whom the screening evaluation suggests a pathological cause, not for the routine confirmation of a normal variant. Growth hormone stimulation testing — using agents such as clonidine, glucagon, or arginine, with measurement of the growth hormone response — is indicated when the growth velocity is subnormal or the insulin-like growth factor one is low, to confirm or exclude growth hormone deficiency. It is not needed for a child with a normal velocity and a normal variant picture. An international guideline on genetic testing in short stature recommends reserving chromosomal microarray and genetic testing for children with additional dysmorphic features, intellectual disability, or a pattern suggestive of a monogenic short stature syndrome, rather than applying it routinely — a principle that keeps over-investigation in check. Magnetic resonance imaging of the pituitary is reserved for the child with confirmed growth hormone deficiency, visual symptoms, or other features of a hypothalamic-pituitary lesion. [7] [10]
Management — Resuscitation
There is no resuscitation phase in the management of a normal-variant short stature, because these are not acute or dangerous conditions — the child is well, growing normally, and at no physiological risk. The "resuscitation" equivalent in this context is the timely recognition that the child does not need rescue from a disease they do not have, which means resisting the pressure to over-investigate, over-refer, or over-treat a normal child driven by parental anxiety or by the clinician's own discomfort with the diagnosis. The first management act is therefore the establishment of the diagnosis with a careful history, an accurate growth chart, a bone age, and a focused blood panel, followed by a clear and confident explanation to the family. [1] [4]
The genuine urgency in the differential lies in not missing the pathological causes that present as short stature, and this is the safety function of the workup. A child whose short stature proves on investigation to be hypothyroidism, coeliac disease, growth hormone deficiency, or Turner syndrome needs prompt disease-specific management, and the discipline of completing the focused panel before labelling the child a normal variant is what ensures these are caught. In the adolescent whose delayed puberty is accompanied by distress, the urgency is psychosocial rather than physiological, and it is managed with a sensitive assessment of the impact and the offer of targeted therapy where appropriate. A short course of testosterone can relieve the distress of a boy who is isolated and bullied for his prepubertal appearance, even though it does not change his final height. [9] [11]
Management — Definitive & Stepwise
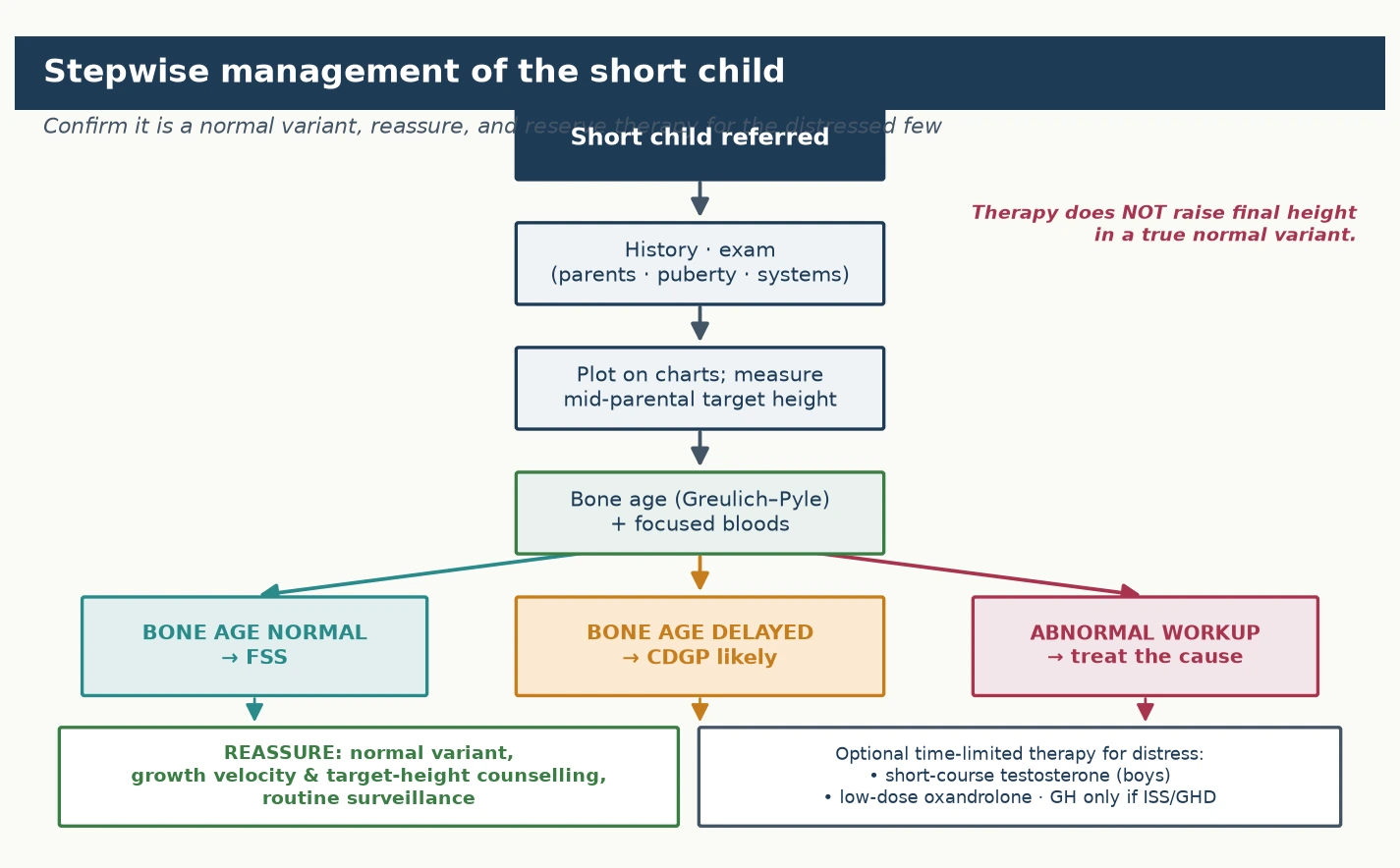
The definitive management of a normal variant is reassurance and surveillance, and the figure lays out the stepwise pathway from referral through diagnosis to the matched management. The guiding principle is that the great majority of children with familial short stature or constitutional delay need no pharmacological therapy at all, because their final height is normal for the family and no available treatment meaningfully raises it. The stepwise approach confirms the variant, excludes the mimics, counsels the family on the expected trajectory, and reserves therapy for the small minority with genuine distress or a confirmed deficiency. [1] [4]
For familial short stature, the management is reassurance, confirmation of the normal trajectory, and routine surveillance. The family is counselled that the child has inherited the family's height, that the growth velocity and bone age are normal, that puberty will proceed on time, and that the final adult height will be short but appropriate for the family and healthy. There is no role for growth hormone therapy in familial short stature, because the growth plate is normal and growth hormone does not raise the final height of a child whose set-point is simply low. Prescribing it exposes the child to years of daily injections, cost, and a small but real burden of adverse effects without a meaningful benefit. Surveillance consists of periodic measurement to confirm that the velocity remains normal and that the child continues to track along the expected trajectory, and the family is given a clear expectation of the adult height range to defuse anxiety. [2] [7]
For constitutional delay, the management is reassurance with the added option of a short course of sex steroid for the distressed adolescent. The family is counselled that the child's biological clock is running late, that puberty will begin spontaneously, and that a late but prolonged growth spurt will carry the child to a normal adult height. Cohort data now corroborate this, showing that boys with constitutional delay enter spontaneous puberty and reach standard adult height without pharmacological therapy. For the adolescent boy distressed by his prepubertal appearance, a short course of monthly intramuscular testosterone for three to six months can accelerate the onset of puberty, relieve the psychosocial burden, and — by accelerating bone-age maturation in step with growth — does not compromise the final height. Low-dose oral oxandrolone is an alternative used in some centres. Growth hormone is not indicated for constitutional delay in the child without deficiency, because it does not increase the final adult height and carries the same cost-and-burden concerns as in familial short stature. [8] [11]
Therapy for the normal variants (specialist-initiated; confirm current dosing)
The psychosocial dimension is central to the management and is often what the family most needs addressed. Short stature and delayed puberty carry a real burden — bullying, social isolation, loss of confidence, and parental anxiety about the child's future — and the clinician who acknowledges this, while firmly and accurately reassuring on the medical trajectory, does more for the child than any prescription. A clear explanation of the growth chart and the expected adult height, written information, a named point of contact, and scheduled review to confirm the trajectory are the elements of good care. Where the psychosocial impact is significant, the short course of testosterone in a distressed boy is a legitimate and effective intervention that targets the distress directly, and it should be offered without the family feeling that they have "failed" the reassurance pathway. [9] [11]
Specific Subtypes & Scenarios
The classic scenario of constitutional delay of growth and puberty in a boy is the one most likely to appear in an examination and the one that best illustrates the diagnostic reasoning. A boy of fourteen presents with no signs of puberty, a height that has drifted down through the centiles in childhood, a bone age of eleven years, and a father who shaved late. The diagnosis is constitutional delay: the growth velocity is normal for the bone age, the height is appropriate for the bone age, and the family history is supportive. The management is reassurance that puberty will begin spontaneously and that the final height will be normal, with the offer of a short course of testosterone if the distress is significant. The teaching point is that the bone age, the family history, and the preserved velocity together make the diagnosis without provocative testing, and that the natural history is reassuring — cohort data confirm that these boys enter spontaneous puberty and reach standard adult height. [9] [11]
The overlap of familial short stature and constitutional delay is a scenario that tests whether the candidate can hold two diagnoses at once. A child of short parents, both of whom were late bloomers, presents short for their age with a delayed bone age and no signs of puberty. The child carries both traits: a low polygenic set-point (familial short stature) and a slowed tempo (constitutional delay). The final height will land at the lower end of the already-short parental target range, and the management is reassurance on both counts, with the realistic expectation that the child will be a short adult. Recognising the overlap prevents the clinician from expecting a late spurt to deliver a normal population height — the target is the family target, not the population mean — and from over-investigating a child who carries two normal traits. [4] [5]
The short girl with a delayed bone age is the scenario that must always include Turner syndrome on the differential. A girl of twelve with short stature, no breast development, and a delayed bone age could have constitutional delay, but she could equally have Turner syndrome, in which short stature is often the dominant or only feature and the classic stigmata may be absent or subtle. The discipline of sending a karyotype or chromosomal microarray in a short girl with delayed puberty and a delayed bone age, before labelling the short stature constitutional, is what prevents the delayed diagnosis of Turner syndrome — a diagnosis that carries important implications for growth hormone therapy, cardiac and renal surveillance, and reproductive counselling. [5] [10]
Numbers worth carrying into the exam
Complications & Pitfalls
The complications of the normal variants are overwhelmingly iatrogenic and psychosocial rather than physiological, and the recognition of this shapes the management. A true normal variant produces a normal adult height and a healthy individual, so the complications arise from the response to the diagnosis rather than from the condition itself. The most common is the psychological burden of short stature and delayed puberty — the bullying, social isolation, and loss of confidence that drive the presentation in the first place — and these are real and deserve active management through counselling, psychosocial support, and, where appropriate, the short course of testosterone. A second category is the burden and risk of unnecessary treatment: years of growth hormone injections for a child who will reach a normal height without them expose the family to cost, daily injections, and a small risk of adverse effects such as intracranial hypertension or glucose intolerance, with no meaningful height benefit. [2] [7]
The most consequential pitfall is mislabelling a pathological process as a normal variant. A child with growth hormone deficiency, hypothyroidism, coeliac disease, or Turner syndrome who is reassured as "just a late bloomer" loses the window for disease-specific therapy — growth hormone works best when started early, hypothyroidism responds promptly to replacement, coeliac disease resolves with a gluten-free diet, and Turner syndrome has a defined growth and surveillance pathway. The safeguard against this pitfall is the disciplined completion of the focused workup before the diagnosis is offered: confirm the growth velocity, read the bone age, run the blood panel, and consider the karyotype in a short girl. A normal variant is a diagnosis of inclusion of the reassuring features and exclusion of the mimics, never a default. [1] [5]
A second pitfall is the reverse error: over-investigating and over-treating a normal child. The pressure from anxious parents, the availability of growth hormone, and the clinician's own discomfort with uncertainty can drive a cascade of provocative tests, imaging, and off-label therapy that medicalises a healthy child. The principle that growth hormone does not raise the final height of a true normal variant, that provocative testing is not needed when the velocity is normal, and that the international genetic-testing guideline reserves testing for children with additional features — all of these guard against the over-medicalisation of normal biology. A third pitfall is confusing constitutional delay with permanent hypogonadotrophic hypogonadism: labelling a Kallmann syndrome patient as a late bloomer delays the recognition and management of a lifelong condition, and the discipline of watching the pubertal tempo and testing when the picture is not resolving is the safeguard. [7] [10]
Prognosis & Disposition
The prognosis of both normal variants is excellent, and this is the single most important fact to convey to the family. Familial short stature delivers a short but healthy adult whose height sits within the mid-parental target range, with normal pubertal development, normal fertility, and a normal lifespan. Constitutional delay delivers a late but ultimately normal puberty and a final adult height within the normal range and the family target range, with no long-term consequence other than the timing. The natural history is so reliably reassuring that cohort data now confirm that boys with constitutional delay reach standard adult height without any pharmacological therapy, and the principal prognostic message is that the condition resolves itself. [8] [11]
The disposition is to routine primary-care surveillance with specialist referral reserved for the atypical or distressed case. The general paediatrician or primary-care clinician confirms the diagnosis, counsels the family, and schedules periodic measurement — typically every six to twelve months — to confirm that the growth velocity remains normal and the trajectory holds. Referral to a paediatric endocrinologist is appropriate when the diagnosis is uncertain, when the growth velocity is subnormal, when there are red flags such as downward centile crossing, when Turner syndrome or another syndrome is suspected, or when the adolescent is significantly distressed and a short course of testosterone is being considered. The endocrinologist's role is to confirm the diagnosis, to run any necessary provocative testing, to initiate the specialist therapy where indicated, and to hand the child back to primary care for ongoing surveillance once the trajectory is secure. [1] [7]
Transition to adult care is relevant chiefly for the child who carries a concurrent diagnosis that persists into adulthood — for example, the child with both constitutional delay and a short adult height who may benefit from adult endocrine input on growth and body image. For the uncomplicated normal variant, the condition resolves with the completion of puberty and no adult follow-up is required. The psychosocial legacy of a difficult adolescence — the bullying and isolation experienced during the years of delayed puberty — may persist, and ensuring that the family has access to psychological support where needed is part of a complete disposition. [9] [11]
Special Populations
Several special populations deserve specific consideration because they reshape the presentation or the management of the normal variants. The adolescent boy distressed by delayed puberty is the population for whom therapy is most often considered, because the psychosocial burden of remaining prepubertal among peers is substantial and the short course of testosterone is an effective, height-neutral intervention. The clinician managing this group must weigh the genuine distress against the principle that most boys need no treatment, offer the family a clear and non-judgemental choice, and ensure that the offer of therapy does not undermine the reassurance about the natural history. The key is to treat the distress, not the height. [9] [11]
Access to publicly funded growth hormone in Australia and New Zealand is governed by strict criteria that generally exclude the normal variants — growth hormone is funded for confirmed growth hormone deficiency, Turner syndrome, chronic kidney disease, Prader-Willi syndrome, and children born small for gestational age who fail to catch up. In selected circumstances it is funded for idiopathic short stature meeting a stringent height threshold. Familial short stature and uncomplicated constitutional delay do not meet these criteria, and the examining clinician should state the local pathway rather than assume a universal standard. The principles are constant: confirm the diagnosis with the growth chart and bone age, exclude the mimics with a focused panel, reserve growth hormone for the funded indications, and run structured surveillance of the growth velocity through primary care. [2] [7]
Socioeconomically disadvantaged, migrant, refugee, and remote populations face additional barriers that shape both the presentation and the management of the normal variants. Growth monitoring depends on regular, accurate measurement, which is harder to sustain across fragmented care, displacement, or geographic isolation, and a child without a reliable growth chart is harder to assess confidently. Nutritional and psychosocial deprivation — themselves more prevalent in disadvantaged populations — are important causes of short stature that must be distinguished from the normal variants, and the clinician should maintain a low threshold to investigate a child whose environment raises the possibility of secondary causes. Equitable access to accurate growth monitoring, to the focused workup, and to specialist referral where indicated is the principle, because the complications of missed pathology fall hardest on the children least able to absorb them. [1] [5]
Evidence, Guidelines & Regional Differences
The evidence base for the normal variants rests on decades of growth research, on the consensus statements that define idiopathic short stature and its boundaries, and on the randomised trials and meta-analyses that have defined the (modest) role of growth hormone. The consensus statement from the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology established the framework for the diagnosis and treatment of idiopathic short stature and clarified its distinction from the normal variants, providing the definitional backbone that clinicians still use. The randomised placebo-controlled trial of growth hormone in peripubertal children with idiopathic short stature, and the subsequent meta-analyses, quantified the modest adult-height gain achievable in that specific population — a gain that does not extend to the normal variants and that must not be extrapolated to them. [2] [3]
Guidelines for the investigation of the short child emphasise the focused, high-yield workup and the avoidance of over-investigation in the clear normal variant. Recent reviews of the evaluation of short and tall stature set out the history, examination, growth-chart, bone-age, and targeted-blood-test approach that secures the diagnosis and excludes the mimics, and the international guideline on genetic testing in short stature reserves chromosomal microarray and genetic testing for children with additional features, keeping the workup proportionate. The management guidelines affirm reassurance and surveillance as the default, with the short course of testosterone for the distressed adolescent boy supported by its established efficacy in accelerating pubertal onset without compromising final height. [1] [10]
Regional differences persist chiefly in the funding and the access criteria for growth hormone, in the structure of paediatric endocrine services, and in the threshold for specialist referral. Growth hormone is an expensive therapy whose public funding is governed by jurisdiction-specific criteria that exclude the normal variants in most regions, and the clinician should ground any discussion of growth hormone in the local criteria rather than in a general assumption of availability. The principles, however, are constant across regions: confirm the diagnosis with the growth chart and bone age, exclude the pathological mimics with a focused panel, reassure and surveil the great majority, reserve sex-steroid therapy for the distressed adolescent, and reserve growth hormone for the funded indications. [7] [8]
Exam Pearls
In a viva, lead with the growth velocity and the trajectory, then state the bone age and the family history, and conclude with the diagnosis and the management plan — that four-step structure answers almost any short-stature question. For a short case, the normal velocity read from the chart, the concordant bone age, the short parents, and the family history of late puberty are the findings that earn the diagnosis. For a long case, the management plan that distinguishes the normal variant from the pathological mimics, that excludes the mimics with a focused panel, and that reserves therapy for the distressed or the deficient is what distinguishes a pass from a commendation. Remember that the bone age is the discriminator between the two variants, and that the growth velocity is the discriminator between a normal variant and disease. [4] [9]
The single most testable principle is that familial short stature and constitutional delay are normal variants with a preserved growth velocity and a normal final height for the family, distinguished from each other by the bone age and the timing of puberty, and distinguished from pathology by the absence of downward centile crossing and the exclusion of the mimics. The management of the great majority is reassurance, not growth hormone. Couple that with the recognition that therapy does not raise the final height of a true normal variant and that the short course of testosterone targets the distress of the delayed boy rather than his height, and you have the framework for the whole topic. Every short-stature question reduces to: is the growth velocity normal, is the bone age concordant, is the family history supportive, and what single pathological mimic must be excluded before reassurance is offered. [1] [7]
References
- [1]Caro R, Savel P, Moss PI. Evaluation of Short and Tall Stature in Children. Am Fam Physician, 2025.PMID 40531152
- [2]Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab, 2008.PMID 18782877
- [3]Leschek EW, Rose SR, Yanovski JA, Troendle JF, Quigley CA, Chipman JJ, et al. Effect of growth hormone treatment on adult height in peripubertal children with idiopathic short stature: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab, 2004.PMID 15240584
- [4]Wit JM, Oostdijk W. Novel approaches to short stature therapy. Best Pract Res Clin Endocrinol Metab, 2015.PMID 26051296
- [5]Stalman SE, Hellinga I, Wit JM, Hennekam RC, van der Kaay DC. Growth failure in adolescents: etiology, the role of pubertal timing and most useful criteria for diagnostic workup. J Pediatr Endocrinol Metab, 2016.PMID 26812776
- [6]Paltoglou G, Dimitropoulos I, Kourlaba G, Chrousos G, Vlachopapadopoulou E. The effect of treatment with recombinant human growth hormone (rhGH) on linear growth and adult height in children with idiopathic short stature (ISS): a systematic review and meta-analysis. J Pediatr Endocrinol Metab, 2020.PMID 33035189
- [7]Danowitz M, Grimberg A. Clinical Indications for Growth Hormone Therapy. Adv Pediatr, 2022.PMID 35985710
- [8]Wit JM. Should Skeletal Maturation Be Manipulated for Extra Height Gain? Front Endocrinol (Lausanne), 2021.PMID 34975773
- [9]Butler G, Purushothaman P. Delayed puberty. Minerva Pediatr, 2020.PMID 32748610
- [10]Dauber A, Jorge AAL, Nilsson O, Backeljauw P, Blair J, Child CJ, et al. International guideline on genetic testing of children with short stature. Eur J Endocrinol, 2026.PMID 41543979
- [11]Luciano TM, Stecchini MF, Antonini SRR. Boys with constitutional delay of growth and puberty developed spontaneous puberty and reached standard adult height without pharmacological therapy. J Pediatr (Rio J), 2025.PMID 40784365