Paeds · endocrinology-diabetes-and-growth
Cushing syndrome in children
Also known as Cushing syndrome · Hypercortisolism · Cushing disease · Paediatric Cushing syndrome · Endogenous hypercortisolism · Iatrogenic Cushing syndrome
Fellowship guide to Cushing syndrome in children: the growth-arrest hallmark that separates it from simple obesity, the confirm-with-two-tests work-up, the ACTH-dependent versus ACTH-independent split, transsphenoidal surgery for pituitary disease, and the perioperative glucocorticoid replacement that prevents adrenal crisis.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single observation that organises everything in paediatric Cushing syndrome is the growth chart decoupling. Cortisol is catabolic and it suppresses the growth-hormone axis, so a child bathed in cortisol gains weight while height falls away. A fat child who keeps growing is almost never Cushing; a fat child who has stopped growing is Cushing until you prove otherwise. [1] [9]
This page covers the recognition and management of Cushing syndrome in children: the growth-arrest presentation, the ACTH-dependent versus ACTH-independent split, the three-test confirmatory work-up, the localising steps including inferior petrosal sinus sampling, transsphenoidal surgery for pituitary disease, adrenalectomy for adrenal disease, and the perioperative glucocorticoid replacement that prevents the adrenal crisis of a suddenly cured child. It links to the adrenal insufficiency leaf for stress-dose detail rather than repeating it. [3] [10]
Overview & Definition
Cushing syndrome means any state of chronic endogenous cortisol excess, whatever the source. The name Cushing disease is reserved for the specific cause — a pituitary corticotroph adenoma that secretes adrenocorticotrophic hormone (ACTH) and drives both adrenal glands to overproduce cortisol. Everything else is Cushing syndrome: adrenal tumours, bilateral adrenal hyperplasia, ectopic ACTH, and the exogenous steroids that cause the commonest form of all. [5]
What makes the paediatric disease distinctive is that children are still growing, and growth is exquisitely sensitive to cortisol. The adult face of Cushing is central obesity, a moon face, striae and hypertension, and those all appear in children too — but the first and most reliable sign is that the child falls off their height centile. Growth arrest is reported in roughly four out of five children with endogenous Cushing, and it is the feature that distinguishes the disease from simple obesity, where height tracks upward with weight. [1] [2]
Classification
Classify Cushing syndrome by where the cortisol signal originates, because the source dictates the investigation pathway and the operation. The figure below splits the syndrome into the exogenous form — overwhelmingly the commonest — and the endogenous causes, then divides endogenous disease into ACTH-dependent and ACTH-independent arms. [3]
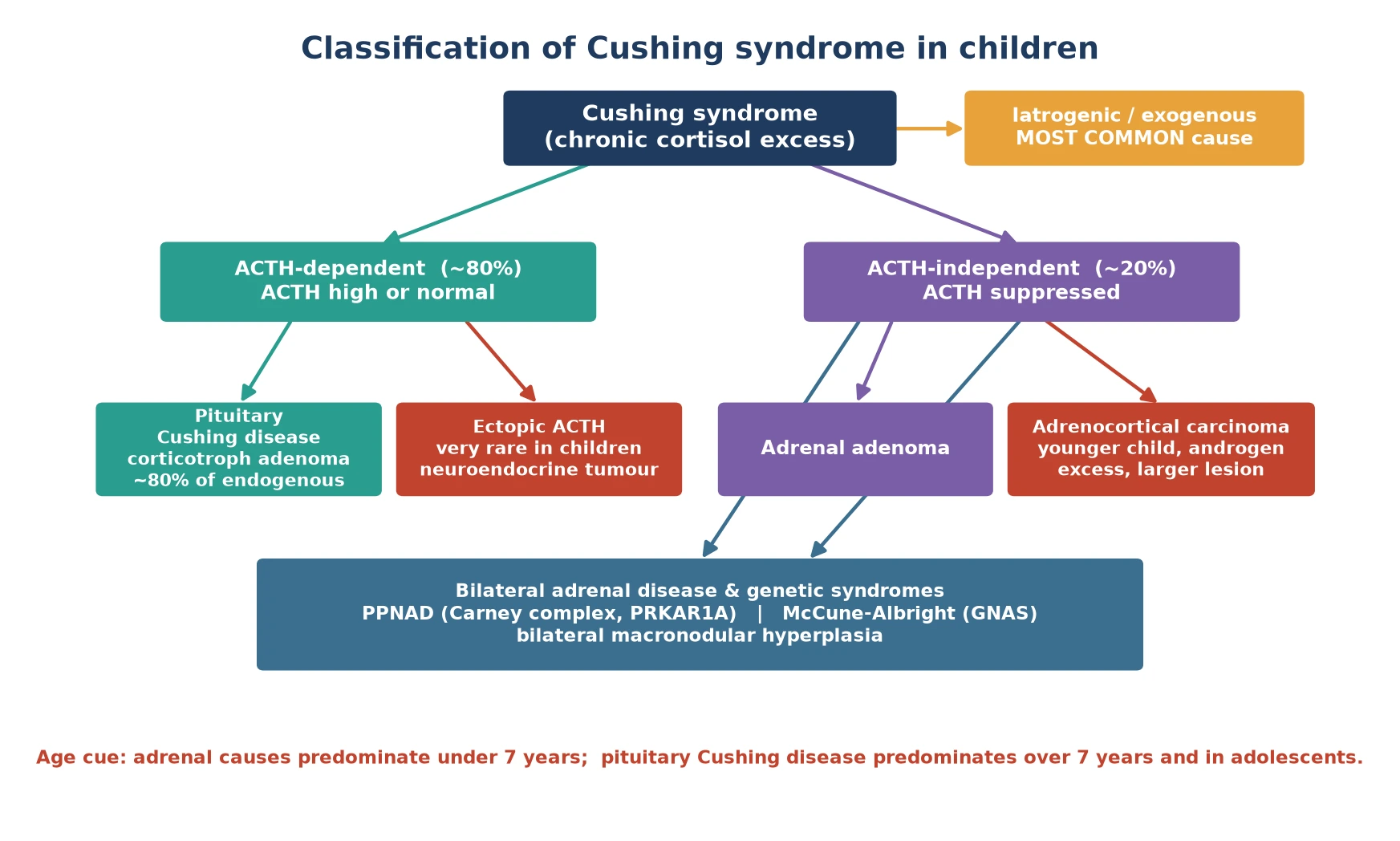
Iatrogenic (exogenous)
- Single most common cause of a Cushingoid child
- Oral, inhaled, intranasal or topical glucocorticoids
- ACTH and endogenous cortisol suppressed (low)
- Cure is to identify and slowly taper the source
ACTH-dependent (~80%)
- Pituitary Cushing disease — corticotroph adenoma, ~80% of endogenous
- Ectopic ACTH — neuroendocrine tumour, very rare in children
- ACTH high or inappropriately normal
- Bilateral adrenal hyperplasia on imaging
ACTH-independent (~20%)
- Adrenal adenoma or adrenocortical carcinoma
- Bilateral disease: PPNAD (Carney complex), McCune-Albright
- ACTH suppressed (low / undetectable)
- Single adrenal mass or bilateral nodularity on imaging
The age of the child reshuffles the probabilities. In children under about seven years, an adrenal cause — adenoma, carcinoma or a genetic bilateral hyperplasia — is the more likely endogenous source. From about seven years onward, and especially in adolescence, pituitary Cushing disease dominates, with a female predominance. Carrying that age cue in mind stops you sending a four-year-old down a pituitary work-up when the answer sits in the adrenal gland. [1] [9]
Epidemiology & Risk Factors
Endogenous Cushing syndrome is rare in children. Population estimates put the incidence of Cushing disease at roughly one to three per million children per year, far below the adult figure, which is why it is a diagnosis of pattern recognition rather than screening. The mean age at paediatric presentation is in the early teenage years, reflecting the dominance of pituitary disease in adolescents. [1] [9]
The strongest risk factor is a hereditary tumour predisposition syndrome. A child with Carney complex (PRKAR1A) may develop primary pigmented nodular adrenocortical disease; a child with McCune-Albright syndrome (GNAS) may develop autonomous adrenal hypercortisolism; and Li-Fraumeni syndrome (TP53) raises the risk of adrenocortical carcinoma, particularly in young children. A family history of early-onset endocrine tumours should lower the threshold to test an atypical, bilateral or very young presentation. [5] [10]
CUSHING
Visceral fat, moon face, supraclavicular and dorsocervical fat pads
24-hour urinary free cortisol is one of the three confirmatory tests
Purple striae wider than 1 cm, easy bruising, thin skin, acne
Growth velocity falls — the paediatric hallmark and key discriminator from obesity
Hypertension in roughly half to two-thirds of paediatric cases
Loss of diurnal rhythm — midnight cortisol stays elevated
Impaired glucose tolerance, emotional lability, school performance decline
The reason exogenous steroid exposure tops the list is its sheer availability — inhaled corticosteroids for asthma, intranasal steroids for allergic rhinitis, potent topical steroids for eczema, and oral or enteral steroids for inflammatory disease. A careful drug history, including over-the-counter and complementary preparations, is the first investigation of any Cushingoid child, because finding the source is the cure. [4]
Pathophysiology
To see why cortisol runs out of control, picture the hypothalamic-pituitary-adrenal axis as a thermostat. Cortisol released from the adrenal cortex normally feeds back to the pituitary and hypothalamus to switch off ACTH, keeping the cortisol level in range and dipping it overnight. A corticotroph adenoma is a rogue thermostat: it secretes ACTH without listening to the feedback, so both adrenal glands are driven to hyperplasia and flood the system with cortisol. [5]
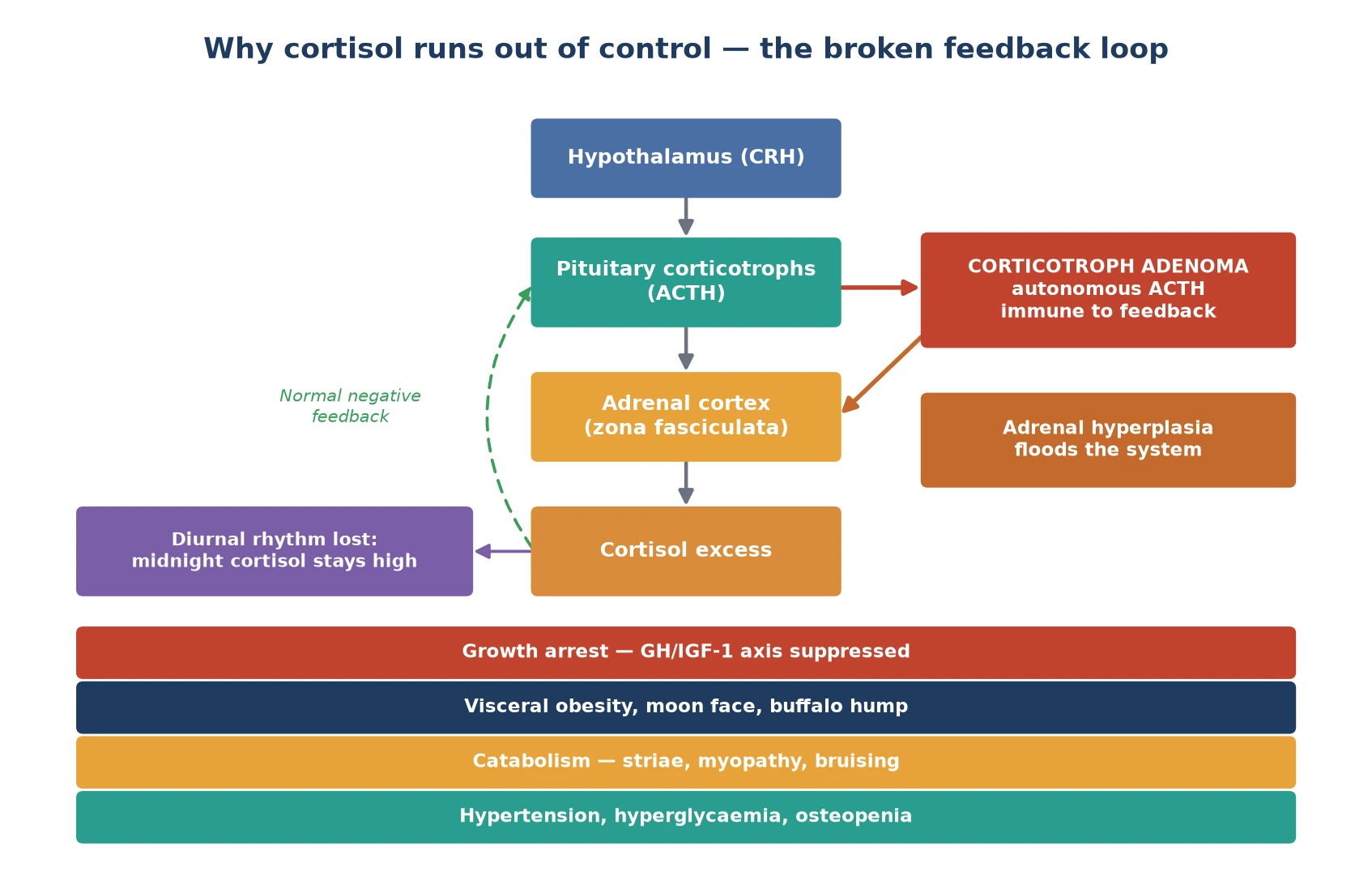
Two features of that broken loop matter at the bedside. First, the normal overnight fall in cortisol is lost, which is why a midnight cortisol is the most sensitive single test — the value that should be near zero stays high. Second, cortisol is a potent catabolic and anti-insulin hormone, so the child loses protein from skin and muscle (striae, bruising, myopathy) while laying down visceral fat and developing hypertension and glucose intolerance. The growth-hormone axis is suppressed, which is the mechanism behind the growth arrest. [1] [5]
The ACTH-independent causes flip the loop. A primary adrenal adenoma or carcinoma makes cortisol on its own, so the pituitary is appropriately switched off and ACTH falls to low or undetectable. The same downstream catabolic and metabolic effects follow, but the adrenal — not the axis — is the lesion. Recognising whether ACTH is high or low is the fork that sends you either to pituitary imaging or to adrenal imaging. [3]
Clinical Presentation
A child with Cushing syndrome usually presents with a slow, insidious change in body shape and behaviour that parents date back several months. The face rounds, the trunk thickens, a fat pad appears between the shoulders and above the collarbones, and the weight climbs. What does not climb is the height — and the growth chart often tells the story before any blood test does, with height crossing centiles downward while weight rises. [1] [2]
The skin carries some of the most specific signs. Striae in Cushing are purplish, broad (wider than one centimetre) and lie over the abdomen, hips, thighs and axillae; they are the colour of unopposed cortisol acting on skin that has lost its collagen. The thin pale or silvery striae of rapid adolescent growth or simple obesity are a different animal. Easy bruising, thin shiny skin and acne add to the cortisol fingerprint. [5]
The metabolic and neuropsychiatric features complete the picture. Hypertension is common, present in roughly half to two-thirds of children, driven by cortisol's mineralocorticoid-like effects and volume expansion. Puberty is often delayed, glucose tolerance is impaired, and many children describe fatigue, emotional lability, irritability or a drop in school performance. Proximal muscle weakness — difficulty climbing stairs or rising from the floor — reflects the catabolic myopathy and is a useful functional sign. [7] [9]
Differential Diagnosis
The differential turns on separating true endogenous Cushing from its mimics, and the growth chart is again the first tool. Simple obesity is the commonest mimic: a heavy child with central fat and striae, but with preserved or accelerated height velocity and a normal overnight cortisol. A child who keeps growing is overwhelmingly likely to have simple obesity, not Cushing. [3]
Points to Cushing
- Height velocity falling while weight rises
- Purple striae wider than 1 cm, easy bruising, thin skin
- Hypertension, proximal myopathy, delayed puberty
- Mood change and school decline out of proportion
- Loss of overnight cortisol dip on testing
Points to a mimic
- Simple obesity: normal or tall stature, advanced bone age
- Exogenous steroid exposure: suppressed ACTH, drug history
- Pseudo-Cushing from severe depression or alcohol (rare in children)
- PCOS: oligomenorrhoea and androgen excess without cortisol excess
- Glucocorticoid resistance: high cortisol, no Cushingoid features
A careful drug history is the second discriminator, because exogenous glucocorticoids reproduce the entire syndrome and are far commoner than any endogenous cause. Ask specifically about inhaled, intranasal, topical and oral steroids, including prescribed and over-the-counter preparations. Glucocorticoid resistance (a glucocorticoid-receptor defect) produces high cortisol levels without the Cushingoid phenotype, and pseudo-Cushing states — major depression, chronic alcohol misuse — can mildly derange cortisol testing, though these are rare in children and usually resolve when the underlying illness is treated. [4] [5]
Clinical & Bedside Assessment
The focused assessment of a suspected Cushingoid child rests on three measurements that take minutes but settle the question: plot the growth, take the blood pressure, and examine the skin. Pull out the growth chart and look for the decoupling — height crossing down, weight crossing up — over the preceding six to twelve months. A fall in height velocity in a child gaining weight is the single most powerful bedside clue. [1]
Measure the blood pressure properly, with an appropriate cuff and, if elevated, four-limb pressures to exclude coarctation. Hypertension in a child is never physiological, and its combination with centripetal obesity and striae points straight at cortisol excess. Examine the skin for the broad purple striae, easy bruising and thin shiny skin, and note the fat distribution — the round face, the supraclavicular fullness, the dorsocervical pad. Look at pubertal staging, which is often delayed, and test proximal muscle power by asking the child to stand from a squat or climb a step. [7] [9]
The assessment is rounded out by asking about mood, sleep and school. Parents often describe a personality change — irritability, tearfulness, withdrawal — that predates the physical signs, and a careful functional history captures the catabolic myopathy and the neuropsychiatric burden that the examination alone may miss. These features, taken with the growth chart, justify moving to the confirmatory biochemical tests rather than re-measuring weight. [2]
Investigations
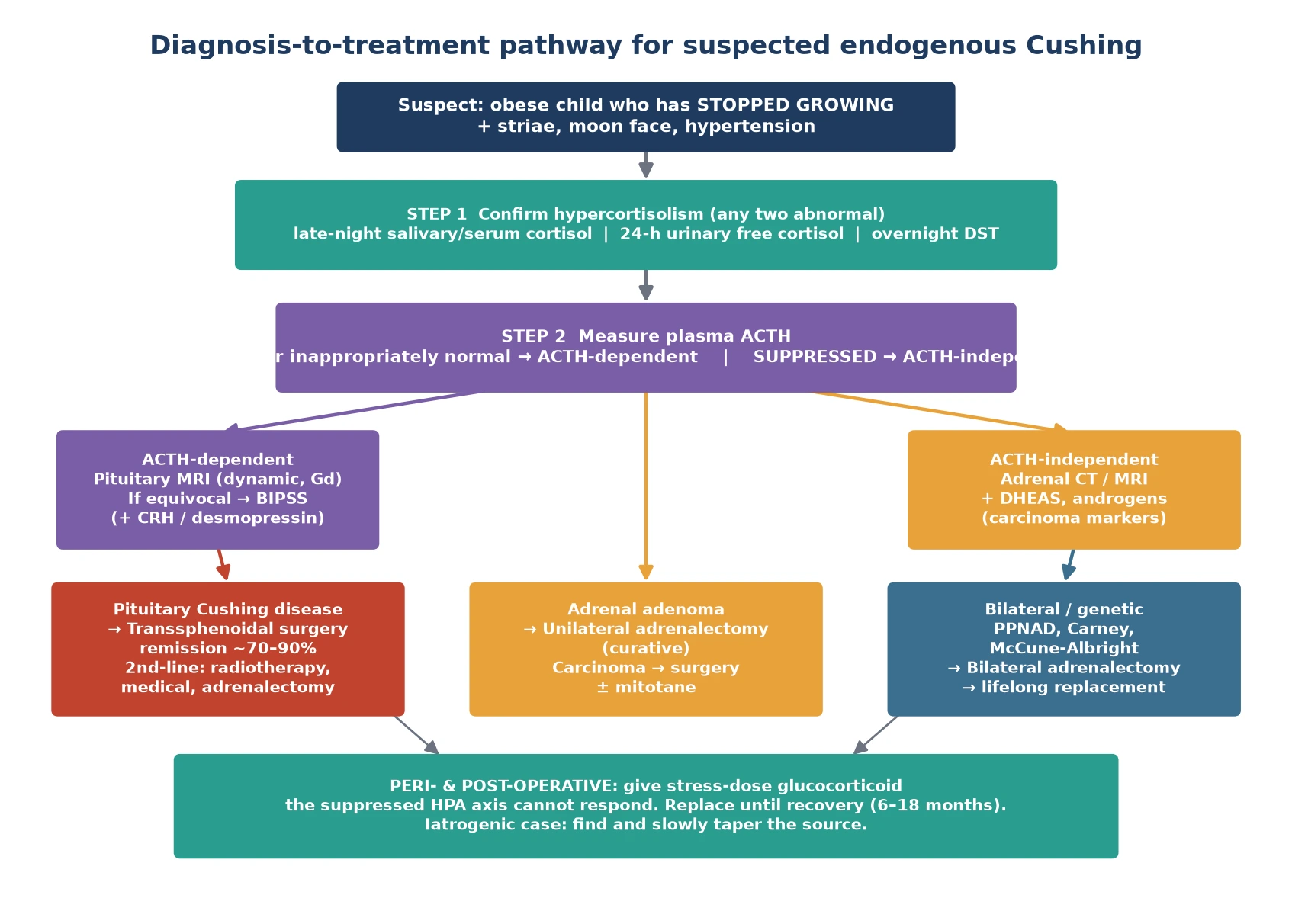
The Endocrine Society diagnosis pathway proceeds in two stages: first confirm that there is genuine cortisol excess, then find its source. Confirm with at least two of three first-line tests, because no single test is perfect and Cushing is cyclic. The three tests are a late-night (midnight) cortisol, a 24-hour urinary free cortisol, and an overnight dexamethasone suppression test. [3]
Late-night cortisol
- Most sensitive single test — loss of diurnal rhythm is earliest change
- Salivary cortisol preferred in children (non-invasive, done at home asleep)
- A sleeping midnight serum cortisol under 50 nmol/L (1.8 µg/dL) argues against Cushing
- Repeat, because the disease is cyclic
24-hour urinary free cortisol
- Integrated measure of the day's cortisol output
- Repeat two to three times; collect must be complete
- Useful for severity and for following response
- Confounded by incomplete collection or high urine volume
Overnight dexamethasone suppression
- Dexamethasone 15 µg/kg (cap 1 mg) at 11 pm; cortisol at 8 am
- Cortisol not suppressed below 1.8 µg/dL (50 nmol/L) supports Cushing
- Dexamethasone is chosen because it does not cross-react in cortisol assays
- False positives with depression, obesity, oestrogen intake
Once cortisol excess is confirmed, measure plasma ACTH to localise. A high or inappropriately normal ACTH means ACTH-dependent disease — pituitary or, rarely, ectopic. A suppressed, low or undetectable ACTH means ACTH-independent disease and points to an adrenal source. This single result sends you down one of two distinct imaging pathways. [3] [5]
For ACTH-dependent disease, image the pituitary with a dynamic gadolinium-enhanced MRI. Many corticotroph adenomas are small, so a normal-looking MRI does not exclude Cushing disease. When the MRI is negative or equivocal, bilateral inferior petrosal sinus sampling is the gold standard: it confirms a pituitary source and helps lateralise the adenoma. A central-to-peripheral ACTH ratio above two at baseline, or above three after corticotrophin-releasing hormone (CRH) stimulation, confirms pituitary Cushing disease. [3] [10]
For ACTH-independent disease, image the adrenae with CT or MRI. A single unilateral mass suggests an adenoma or carcinoma — and a larger lesion (over four centimetres), androgen excess and a high DHEAS point toward carcinoma. Bilateral nodularity or the small pigmented nodules of primary pigmented nodular adrenocortical disease suggest a genetic syndrome, which should prompt screening for Carney complex or McCune-Albright and referral to a clinical genetics service. [5] [9]
Management — Resuscitation
Cushing syndrome is rarely a time-critical resuscitation emergency in the way adrenal crisis is, but two acute situations demand immediate action. The first is severe hypercortisolism with dangerous hypokalaemia, hypertension or psychosis; the second, and far commoner, is the adrenally insufficient child who has just been cured. Recognising both is the resuscitation skill. [4]
When a child is cured of Cushing — by transsphenoidal surgery, adrenalectomy or tapering of exogenous steroid — the suppressed pituitary-adrenal axis cannot generate cortisol, so the child tips into adrenal insufficiency within hours to days. Treat this exactly as adrenal crisis: give a stress dose of hydrocortisone, intravenously in the perioperative period, alongside intravenous fluids and glucose as needed. Do not wait for a cortisol level in a child who is hypotensive, hyponatraemic or hypoglycaemic after curative surgery. [4]
For severe endogenous hypercortisolism awaiting definitive surgery — especially with marked hypokalaemia, hypertension or neuropsychiatric breakdown — a steroidogenesis inhibitor such as ketoconazole or metyrapone can lower cortisol as a bridge, managed jointly with paediatric endocrinology. Control the blood pressure, correct the potassium, and watch the glucose, but recognise that definitive cure is surgical and the medical therapy is a temporising measure. [4] [5]
Management — Definitive & Stepwise
Definitive treatment is dictated by the source. For pituitary Cushing disease, first-line treatment is transsphenoidal selective adenomectomy by an experienced pituitary surgeon, which achieves remission in roughly seventy to ninety percent of paediatric cases in expert hands. When no adenoma is found at surgery, a hemi- or total hypophysectomy may be considered. Postoperative cortisol should fall to low levels, confirming cure, and glucocorticoid replacement is then begun. [6] [10]
For adrenal adenoma, unilateral adrenalectomy is curative. For adrenocortical carcinoma, complete surgical resection is the mainstay, with adjuvant mitotane and chemotherapy guided by stage and a multidisciplinary oncology-endocrine team. For bilateral adrenal disease such as primary pigmented nodular adrenocortical disease or McCune-Albright, bilateral adrenalectomy is often required, after which the child has permanent primary adrenal insufficiency and needs lifelong hydrocortisone and fludrocortisone replacement. [4] [5]
When pituitary surgery fails or the disease recurs, the stepwise options are repeat transsphenoidal surgery, pituitary radiotherapy (conventional or stereotactic), medical therapy with steroidogenesis inhibitors or a glucocorticoid-receptor antagonist, and ultimately bilateral adrenalectomy as a last resort — which carries the long-term risk of Nelson syndrome, pituitary corticotroph hyperplasia driven by loss of cortisol feedback. Each step is a multidisciplinary decision weighing latency, side effects and the child's growth. [4] [6]
Monitoring is the long game. Track growth — catch-up growth after cure is expected but final height may still be compromised — and follow cortisol, blood pressure, bone density and glucose. After successful transsphenoidal surgery, the child stays on replacement glucocorticoid until the suppressed axis recovers, often six to eighteen months, guided by morning cortisol or a low-dose ACTH stimulation test. Lifelong endocrine surveillance and a structured transition to adult care complete the pathway. [2] [9]
Specific Subtypes & Scenarios
Pituitary Cushing disease is the dominant endogenous subtype in school-age children and adolescents. A corticotroph microadenoma drives bilateral adrenal hyperplasia, and the presentation is the classic growth arrest with centripetal obesity. Transsphenoidal surgery is first-line, and inferior petrosal sinus sampling is reserved for cases where the pituitary MRI is negative or non-lateralising. [6] [10]
Adrenocortical carcinoma matters disproportionately in young children. It tends to present under five years, often with a larger adrenal mass (over four centimetres), virilisation from androgen co-secretion, and a higher DHEAS. The distinction from a benign adenoma turns on size, imaging features, hormone profile and histology, and management is surgical resection with adjuvant therapy guided by stage. Li-Fraumeni syndrome should be considered in any young child with adrenocortical carcinoma. [5]
Primary pigmented nodular adrenocortical disease (Carney complex) and McCune-Albright syndrome are the genetic bilateral causes. They produce ACTH-independent cortisol excess from both glands, the pituitary ACTH is suppressed, and the adrenal imaging shows bilateral small nodules or enlargement. Carney complex carries extra features — cardiac myxomas, lentigines, other endocrine tumours — that demand their own surveillance. Bilateral adrenalectomy is often the definitive treatment, leaving the child with permanent adrenal insufficiency. [5] [9]
Ectopic ACTH from a neuroendocrine tumour is vanishingly rare in children compared with adults, but it enters the differential of ACTH-dependent disease when the petrosal sinus sampling does not confirm a pituitary source. Iatrogenic Cushing is, in practice, the commonest subtype of all: the child on high-dose inhaled or topical steroids who develops the phenotype, with a suppressed ACTH and a low endogenous cortisol. The treatment is to find the source and taper it slowly, never abruptly, to avoid precipitating adrenal crisis. [4]
Complications & Pitfalls
The untreated disease carries a heavy burden: short adult stature from cortisol-driven skeletal maturation and growth-hormone suppression, osteoporosis with vertebral fracture, persistent hypertension, impaired glucose tolerance or diabetes, and a marked neuropsychiatric toll. Many of these are partly reversible with cure, but final height is the long-term battleground and may remain compromised despite successful treatment. [1] [9]
The treatment carries its own complications. Glucocorticoid over-replacement after cure suppresses the recovering axis and steals growth, so the replacement dose must be physiological, not high. Transsphenoidal surgery risks hypopituitarism, diabetes insipidus and cerebrospinal fluid leak. Bilateral adrenalectomy trades Cushing for permanent adrenal insufficiency and the risk of Nelson syndrome, in which the pituitary corticotrophs hyperplase under unopposed ACTH drive. Each trade-off is a reason for expert, centralised management. [4] [6]
The classic pitfall is the mislabelled obese child. Treating simple obesity as Cushing, or chasing an adrenal tumour in a child on inhaled steroids, wastes time and causes harm — but missing genuine Cushing because the growth chart was not plotted, or attributing the phenotype to "just obesity," is the more dangerous error. Reaching for the growth chart and a drug history before the imaging is the habit that prevents both. [1] [3]
Prognosis & Disposition
With expert transsphenoidal surgery, pituitary Cushing disease remits in roughly seventy to ninety percent of children, and catch-up growth usually follows cure. Final height, however, may still fall short of the genetic target because of the period of cortisol-driven skeletal advancement and growth-hormone suppression before treatment, which is why earlier diagnosis is the single biggest modifiable prognostic factor. [6] [9]
Recurrence occurs in a minority after initial remission, so structured surveillance — periodic cortisol testing and growth monitoring — continues for years. Adrenocortical carcinoma carries a prognosis driven by stage and completeness of resection, and the genetic bilateral causes require lifelong surveillance for their associated tumours. Disposition is to a specialist paediatric endocrinology service with pituitary and adrenal surgical expertise, and a planned transition to adult endocrinology in late adolescence. [10]
Special Populations
Young children (under seven years) are more likely to have an adrenal cause or a genetic syndrome than a pituitary adenoma, so the work-up leans toward adrenal imaging and genetics. Adrenocortical carcinoma in a very young child should prompt consideration of Li-Fraumeni syndrome, and bilateral disease should prompt screening for Carney complex or McCune-Albright. [5] [9]
Iatrogenic Cushing is its own population — the child on inhaled, intranasal, topical or oral glucocorticoids. The treatment is to identify the source and taper it slowly, because an abrupt stop precipitates adrenal crisis in a child whose axis is suppressed. Coordinate the taper with the prescribing team (respiratory, dermatology, rheumatology) and arrange glucocorticoid cover for intercurrent illness during the wean. [4]
Adolescents face the added burden of delayed puberty, body-image distress, menstrual disturbance and mood disorder, and they need honest counselling about fertility, replacement and the long transition off steroids. Indigenous, migrant and remote-dwelling families may face the logistics of accessing a specialist pituitary-adrenal service, so a clear local pathway, telehealth support and a written emergency plan that travels with the child are essential. [2] [10]
Evidence, Guidelines & Regional Differences
The evidence base is anchored by the Endocrine Society's two clinical practice guidelines — the 2008 diagnosis guideline and the 2015 treatment guideline — together with the 2024 consensus on pituitary adenomas in childhood and adolescence. The paediatric-specific evidence draws on the National Institutes of Health cohort described by Magiakou and colleagues, the single-centre surgical series of Kanter and colleagues, and the recent epidemiology and outcome review by Ferrigno and colleagues. [3] [4] [10]
Regional practice differences are modest because the Endocrine Society guidelines are adopted internationally, but the threshold for inferior petrosal sinus sampling, the availability of paediatric pituitary surgeons, and access to medical therapies such as pasireotide or mifepristone vary between centres. In Australia and New Zealand, suspected paediatric Cushing is referred to a tertiary paediatric endocrine service with a linked neurosurgical team, reflecting the centralised expertise that the evidence supports. [5] [10]
The main controversies are the optimal timing and intensity of medical therapy before surgery in severe disease, the role of modified-release hydrocortisone in the post-cure replacement phase, and the long-term management of Nelson syndrome after bilateral adrenalectomy. The evidence in these areas is limited in children and is extrapolated from adult series, so decisions are individualised within a multidisciplinary team. [4] [9]
Exam Pearls
Hold one sentence for the viva: the child who is gaining weight but has stopped growing has Cushing syndrome until proven otherwise, and the investigation confirms cortisol excess with two of three tests before localising by ACTH. The growth chart is the discriminator that separates Cushing from simple obesity, where height tracks upward with weight. [1] [3]
State the frequently misremembered facts correctly — these are the details examiners test. Striae in Cushing are purple and wider than one centimetre, not the thin silver lines of rapid growth. The late-night cortisol is the most sensitive single test because loss of the diurnal rhythm is the earliest change. For the overnight dexamethasone suppression test in children, give dexamethasone 15 µg per kilogram, capped at 1 mg. A central-to-peripheral ACTH ratio above two at baseline, or above three after CRH on inferior petrosal sinus sampling confirms pituitary disease. And the cured child is adrenally insufficient, so stress-dose glucocorticoid is part of every cure. [3] [5]
The high-yield lesion-sign pairings: an obese child with growth arrest and purple striae is Cushing; a young child with a large adrenal mass and virilisation is adrenocortical carcinoma (think Li-Fraumeni); a Cushingoid child with a clear steroid exposure history has iatrogenic disease; and bilateral adrenal nodularity with skin pigmentation and myxomas is Carney complex. These pairings do most of the diagnostic work in the short case. [5] [9]
References
- [1]Magiakou MA; Mastorakos G; Oldfield EH; et al Cushing's syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med, 1994.PMID 8052272
- [2]Magiakou MA; Chrousos GP Cushing's syndrome in children and adolescents: current diagnostic and therapeutic strategies. J Endocrinol Invest, 2002.PMID 11929092
- [3]Nieman LK; Biller BM; Findling JW; et al The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2008.PMID 18334580
- [4]Nieman LK; Biller BM; Findling JW; et al Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2015.PMID 26222757
- [5]Lacroix A; Feelders RA; Stratakis CA; Nieman LK Cushing's syndrome. Lancet, 2015.PMID 26004339
- [6]Kanter AS; Diallo AO; Jane JA Jr; et al Single-center experience with pediatric Cushing's disease. J Neurosurg, 2005.PMID 16302612
- [7]Magiakou MA; Smyrnaki P; Chrousos GP Hypertension in Cushing's syndrome. Best Pract Res Clin Endocrinol Metab, 2006.PMID 16980206
- [8]Stratakis CA; Mastorakos G; Magiakou MA; et al Thyroid function in children with Cushing's disease before and after transsphenoidal surgery. J Pediatr, 1997.PMID 9427898
- [9]Ferrigno R; Hasenmajer V; Caiulo S; et al Paediatric Cushing's disease: Epidemiology, pathogenesis, clinical management and outcome. Rev Endocr Metab Disord, 2021.PMID 33515368
- [10]Korbonits M; Blair JC; Boguslawska A; et al Consensus guideline for the diagnosis and management of pituitary adenomas in childhood and adolescence: Part 2, specific diseases. Nat Rev Endocrinol, 2024.PMID 38336898