Paeds · endocrinology-diabetes-and-growth
Delayed puberty and hypogonadism in adolescents
Also known as Constitutional delay of growth and puberty · CDGP · Delayed puberty · Hypogonadotropic hypogonadism · Congenital hypogonadotropic hypogonadism · CHH · Kallmann syndrome · Hypergonadotropic hypogonadism · Primary gonadal failure
A fellowship approach to the adolescent whose puberty is late or has stalled: confirm the timing against sex-specific thresholds (no testicular enlargement by 14 in boys, no thelarche by 13 in girls, or a pause over 2 years), split the differential with gonadotrophins into central versus gonadal causes, separate the common and benign constitutional delay of growth and puberty from permanent congenital hypogonadotropic hypogonadism, and treat by reassuring the temporary, inducing puberty in the permanent, and replacing sex steroids for life in primary gonadal failure.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who works through three layers in order. The first is the timing: has puberty genuinely failed to start, or has it started and then stalled, and where does this adolescent sit against the sex-specific thresholds. The second is the axis: a morning testosterone or oestradiol with gonadotrophins, read against bone age and growth velocity, tells you whether the problem is central or gonadal. The third is the clock versus the furnace: constitutional delay is a late but intact clock that will start on its own, whereas congenital hypogonadotropic hypogonadism is a permanently defective clock that needs exogenous sex steroids to drive puberty, and primary gonadal failure is a clock with no furnace to drive. The candidate who can name the anosmia of Kallmann, the streak ovaries of Turner, and the small firm testes of Klinefelter, and who knows when to order a pituitary MRI, passes the station. [1] [10]
Overview & Definition
Delayed puberty is defined by the failure of secondary sexual characteristics to appear within two standard deviations of the population mean. In practical terms this means no testicular enlargement — a testicular volume below four millilitres, measured with a Prader orchidometer — by 14 years in boys, or no breast budding (thelarche) by 13 years in girls. A second, equally important definition is a stall in progression: more than two years between the onset of puberty and the next stage, or more than five years between thelarche and menarche in girls. The timing thresholds matter because puberty begins with reactivation of the hypothalamic gonadotropin-releasing hormone pulse generator, which has been suppressed since the neonatal minipuberty, and a delay signals either a late but intact generator or a permanent defect somewhere in the hypothalamic-pituitary-gonadal axis. [1] [10]
Classification
The clinically useful classification rests on the gonadotrophin level, because it tells you where the block sits in the axis. Low or inappropriately normal gonadotrophins define the hypogonadotropic group, in which the problem is central — the hypothalamus is not generating enough gonadotropin-releasing hormone, or the pituitary is not responding. High gonadotrophins define the hypergonadotropic group, in which the problem is the gonad — it has failed, the negative feedback is lost, and the pituitary drives the gonadotrophins up in a futile attempt to stimulate it. This single split, made with a morning blood sample, converts an undifferentiated clinical picture into a focused investigation plan. [1] [5]
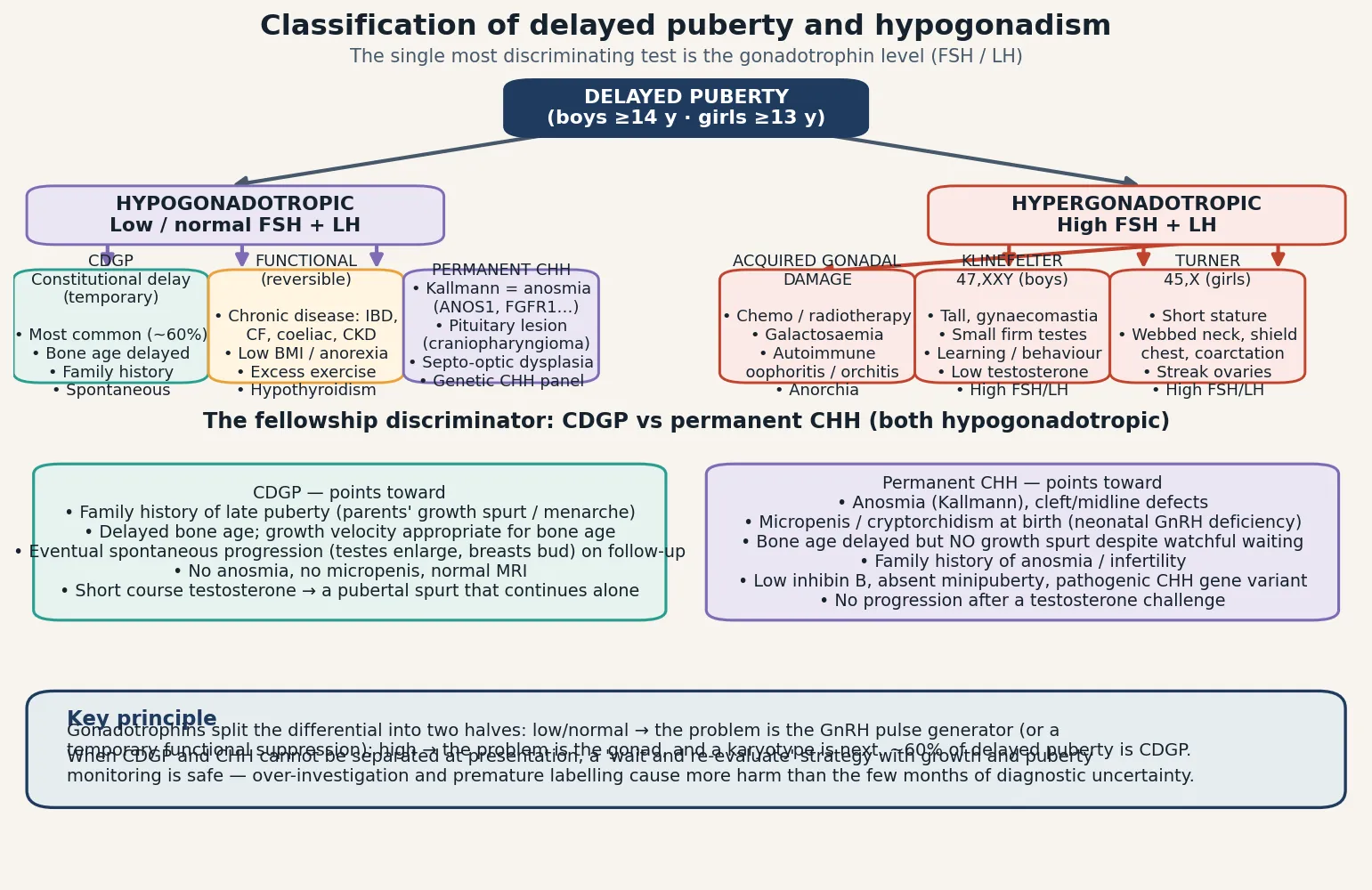
Within the hypogonadotropic group sit three categories. Constitutional delay of growth and puberty is a temporary late-running clock, the commonest cause of delayed puberty in boys, with a strong family history and delayed bone age. Functional hypogonadotropic hypogonadism is a reversible suppression of the GnRH pulse generator by undernutrition, chronic systemic disease, excessive exercise, hypothyroidism, or hyperprolactinaemia. Permanent congenital hypogonadotropic hypogonadism is an irreversibly defective GnRH system — the olfactory (Kallmann) and normosmic forms of CHH, plus structural hypothalamic-pituitary lesions such as craniopharyngioma. The hypergonadotropic group comprises the primary gonadal failures: Klinefelter syndrome, Turner syndrome, and acquired damage from gonadotoxic treatment, galactosaemia, or autoimmune gonaditis. [1] [11]
Epidemiology & Risk Factors
The epidemiology of delayed puberty is dominated by the sheer frequency of constitutional delay, particularly in boys. Sedlmeyer and Palmert's analysis of a large academic case series found that constitutional delay of growth and puberty accounted for roughly 60 percent of boys and 30 percent of girls referred for delayed puberty evaluation — a sex skew that reflects both physiology (boys have a wider normal range of pubertal timing) and referral patterns (short, immature boys are brought to attention more readily than girls of similar timing). The remainder is split between functional suppression, permanent central causes, and primary gonadal failure. [2] [11]
The strongest risk factor for constitutional delay is a family history of late puberty. Sedlmeyer, Hirschhorn and Palmert's pedigree analysis showed that constitutional delay aggregates strongly within families, and the inheritance patterns are complex — consistent with autosomal dominant with incomplete penetrance in some kindreds, and polygenic in others. Wehkalampi and colleagues confirmed high familial aggregation in both boys and girls referred to specialist care. Asking the parents directly is high-yield: when did the father have his growth spurt, when did the mother reach menarche, were they the shortest in their class until late. A parent who "was always the smallest until age sixteen" makes constitutional delay far more likely. [3] [4]
The risk factors for permanent hypogonadism are different. A family history of anosmia or infertility, midline birth defects (cleft lip or palate, single central incisor), and syndromic features point to congenital hypogonadotropic hypogonadism. A history of gonadotoxic treatment — alkylating agents, platinum compounds, total-body irradiation, testicular or ovarian radiation — makes primary gonadal failure likely in an oncology survivor. Autoimmune disease, Turner or Klinefelter stigmata on examination, and galactosaemia raise the pre-test probability for the gonadal-failure group. Chronic illness — inflammatory bowel disease, cystic fibrosis, coeliac disease, chronic kidney disease, anorexia nervosa — is the engine of the functional group. [5] [10]
Pathophysiology
Puberty begins when the hypothalamic gonadotropin-releasing hormone pulse generator is reactivated after a childhood quiescence. Kisspeptin neurons in the arcuate nucleus drive pulsatile GnRH release into the hypophyseal portal system, the anterior pituitary responds with pulses of luteinising hormone and follicle-stimulating hormone, and the gonad is stimulated to produce sex steroids (testosterone from Leydig cells in boys, oestradiol from the ovary in girls) and gametes. The sex steroids drive the physical changes of puberty and feed back negatively on the hypothalamus and pituitary to close the loop. The timing of reactivation is set by an interaction of genetic background, body composition (leptin is a permissive signal), and environmental factors, which is why constitutional delay runs in families and why undernutrition suppresses the axis. [1] [5]

A block at the hypothalamus or pituitary removes the trophic drive to the gonad, so the gonad produces little or no sex steroid and the gonadotrophins remain low or inappropriately normal — the hypogonadotropic pattern. Constitutional delay is the temporary version of this: the pulse generator is intact but starts late. Functional suppression is the reversible version: illness or undernutrition dampens the pulse amplitude. Congenital hypogonadotropic hypogonadism is the permanent version: the GnRH neurons are absent or defective, classically because they never migrated from the olfactory placode (Kallmann syndrome). A block at the gonad itself destroys the steroid-producing cells, the negative feedback is lost, and the gonadotrophins climb in a vain attempt to drive the unresponsive gonad — the hypergonadotropic pattern of Klinefelter, Turner and acquired gonadal failure. [5] [6]
Two developmental details sharpen the bedside reasoning. First, the neonatal minipuberty: a transient GnRH surge in the first six months of life activates the axis and produces measurable testosterone in male infants. Permanent CHH is already evident in this window — a boy with micropenis and undescended testes at birth, or a history of low neonatal testosterone, pushes the diagnosis away from constitutional delay and toward CHH. Second, the bone-mass cost: sex steroids drive the rapid skeletal mineralisation of adolescence, and every year of untreated hypogonadism forfeits peak bone mass. Pubertal induction is therefore not a cosmetic intervention but a strategy to protect the skeleton for life. [9] [12]
Clinical Presentation
The presentation is usually an adolescent, more often a boy, brought by parents concerned about short stature, immaturity relative to peers, or simply the absence of the changes they expected by this age. Constitutional delay presents with a consistent triad: short stature through childhood with a normal growth velocity for bone age, a delayed bone age that matches the height age, and a family history of late puberty. The adolescent is otherwise well, has no dysmorphic features, no chronic illness, and a normal sense of smell. The growth chart shows a child who has tracked along a lower centile line throughout childhood with a typical prepubertal deceleration, and the bone age lags the chronological age by two years or more. [1] [2]
Permanent congenital hypogonadotropic hypogonadism presents with complete absence of puberty, often with a standing height that is normal or even tall for age (because the pubertal growth spurt has not fused the growth plates), and the discriminating features are anosmia, midline defects, or a history of micropenis and cryptorchidism. The bone age is delayed but there is no growth spurt on follow-up, in contrast to constitutional delay where a spurt eventually appears. Klinefelter syndrome presents in a boy with tall stature, long limbs, small firm testes (typically under six millilitres), gynaecomastia, and learning or behavioural vulnerabilities, though many cases are not diagnosed until adulthood infertility evaluation. Turner syndrome presents in a girl with short stature, webbed neck, shield chest, cubitus valgus, and often a cardiac anomaly such as coarctation of the aorta. [6] [7] [8]
Functional hypothalamic suppression presents in the context of the cause: an intense female athlete or dancer with low body mass and secondary amenorrhoea or primary amenorrhoea (the female athlete triad of low energy availability, menstrual dysfunction, and low bone density), an adolescent with anorexia nervosa, or a child with poorly controlled inflammatory bowel disease, cystic fibrosis, or coeliac disease. The key is that the suppression tracks the underlying state — improving nutrition and reducing the training load, or controlling the disease, restores the axis. Recognising this group matters because the treatment is not sex steroids but reversal of the precipitant. [10] [11]
Differential Diagnosis
The differential is organised by the gonadotrophin result. In the hypogonadotropic group the competitors are constitutional delay, functional suppression, and permanent central causes. The structural central causes are the ones not to miss: a craniopharyngioma or other hypothalamic-pituitary tumour can present with delayed puberty, growth failure, visual-field defects, headache, or diabetes insipidus, and any adolescent with these features needs a pituitary MRI. Infiltrative disease (Langerhans cell histiocytosis), genetic syndromes (septo-optic dysplasia, Prader-Willi), and isolated growth hormone deficiency also enter the central differential. The separation between constitutional delay and CHH is the classic fellowship problem, and at first presentation it is sometimes genuinely impossible — a period of watchful waiting with growth and pubertal monitoring is both safe and appropriate. [1] [5]
In the hypergonadotropic group the differential is the causes of primary gonadal failure. Klinefelter syndrome (47,XXY) is the commonest genetic cause in boys, with an incidence of roughly one in 600 male births. Turner syndrome (45,X or mosaic) affects roughly one in 2500 girls and is the leading explanation for hypergonadotropic hypogonadism in a short girl. Acquired causes include gonadotoxic chemotherapy and radiation (dose-dependent, with alkylating agents and testicular radiation the most damaging), galactosaemia, autoimmune ovarian or testicular failure (often in autoimmune polyglandular syndromes), and traumatic or surgical loss. Bilateral cryptorchidism or anorchia (the vanishing testes syndrome) present with absent puberty and high gonadotrophins. [7] [8]
Clinical & Bedside Assessment
The assessment is an outpatient exercise built around the growth chart, the pubertal examination, and a directed history. Begin with the growth trajectory: plot every available height and weight, calculate the current growth velocity, and compare the height centile with the mid-parental target height. In constitutional delay the child has tracked along a lower centile throughout childhood, the growth velocity is normal for bone age, and the bone age (a radiograph of the left hand and wrist, read by Greulich-Pyle or Tanner-Whitehouse standards) lags the chronological age by two years or more and matches the height age. A child whose height has fallen away from the centiles, or whose growth velocity has dropped below four centimetres per year, has something other than simple constitutional delay and needs fuller investigation. [1] [10]
The pubertal examination uses Tanner staging. In boys, measure testicular volume with a Prader orchidometer: a volume of four millilitres or a long axis of 2.5 centimetres marks the onset of puberty, and a volume persistently below four millilitres at 14 defines delayed puberty. Note the consistency (firm and small in Klinefelter), the position (undescended testes point to CHH or anorchia), and the penile size (micropenis suggests neonatal GnRH deficiency). In girls, stage breast development (thelarche is usually the first sign) and pubic hair separately, and assess for thelarche variant. A pelvic ultrasound assesses the uterus and ovaries when gonadal dysgenesis is suspected. Look systematically for the stigmata of Turner (webbed neck, shield chest, cubitus valgus, cardiac murmur of coarctation), Klinefelter (tall stature, gynaecomastia, small firm testes), and Noonan (which phenocopies Turner in a boy). [10] [8]
The history must include the parents' pubertal timing (father's growth spurt age, mother's menarche age), a three-generation pedigree for anosmia, infertility, and consanguinity, the smell test (anosmia is the key to Kallmann), and a search for chronic disease (gut symptoms for inflammatory bowel disease and coeliac disease, respiratory for cystic fibrosis, exercise and eating patterns for the athlete triad). Ask about headaches, visual symptoms, and polyuria-polydipsia to exclude a hypothalamic-pituitary lesion, and take a detailed treatment history for gonadotoxic exposure. The bedside assessment converts directly into the investigation plan: a normal, well, short-for-age boy with a delayed bone age and a positive family history needs minimal investigation, while any adolescent with red-flag features needs an endocrine panel and imaging. [11] [5]
Investigations
The first-tier investigation is the hormonal panel: a morning (around 8 a.m., when testosterone peaks) testosterone in boys or oestradiol in girls, with luteinising hormone, follicle-stimulating hormone, thyroid function, and prolactin. The gonadotrophin level is read in relation to the sex steroid: low or inappropriately normal gonadotrophins with a low sex steroid confirm a central (hypogonadotropic) problem; high gonadotrophins with a low sex steroid confirm primary gonadal failure. A high prolactin, a low free thyroxine, or markers of systemic disease refine the functional causes. The bone age radiograph is performed in every case and interpreted alongside the growth chart. [1] [10]
When the gonadotrophins are high, a karyotype (or chromosomal microarray) is the next step to confirm Klinefelter (47,XXY) or Turner (45,X or mosaic). Pelvic or testicular ultrasonography characterises the gonads (streak ovaries in Turner, small testes in Klinefelter, absence of testes in anorchia). Anti-Müllerian hormone and inhibin B are markers of gonadal reserve that can refine the picture, particularly in oncology survivors and in distinguishing isolated gonadotropin deficiency from constitutional delay. When the picture is central and the cause is not obviously constitutional or functional, a pituitary MRI excludes a structural lesion — the threshold for imaging is lower when there are headaches, visual symptoms, growth arrest, diabetes insipidus, or a high prolactin, and higher in the classic well constitutional-delay boy. [5] [8]
When CDGP and CHH cannot be separated at the first visit
At the first presentation, the well boy with delayed bone age, absent puberty, and a borderline family history may sit in genuine diagnostic uncertainty between constitutional delay and permanent congenital hypogonadotropic hypogonadism. There is no single test that cleanly separates them at that moment — even the GnRH stimulation test and the kisspeptin response overlap. The safe and evidence-based strategy is a structured period of watchful waiting with three- to six-monthly assessment of growth and pubertal staging, combined with a short therapeutic trial of testosterone if the psychological burden is high. Constitutional delay will show a pubertal response that continues after the testosterone stops; CHH will not. Premature labelling causes more harm than a few months of careful observation. [1] [12]
Genetic testing has an increasing role. A CHH gene panel (ANOS1, FGFR1, GNRHR, TAC3, TACR3, CHD7 and many others) is offered when the clinical picture suggests permanent central hypogonadism, particularly with anosmia or midline defects. CHD7 mutations cause CHARGE syndrome and a CHH phenotype. A negative genetic panel does not exclude CHH, because the oligogenic inheritance means many pathogenic combinations remain unidentified — genetic testing supports but does not rule out the diagnosis. Smell testing (the University of Pennsylvania Smell Identification Test or simpler bedside alternatives) documents the anosmia that defines Kallmann syndrome. [5] [6]
Management — Resuscitation
Delayed puberty is not a resuscitation diagnosis, but the first clinical priority is to exclude the time-critical central lesions before settling into outpatient management. An adolescent with delayed puberty plus headache, visual-field loss, new growth arrest, or polyuria-polydipsia has a potential hypothalamic-pituitary tumour (craniopharyngioma, germinoma, prolactinoma) and needs a pituitary MRI on an urgent rather than routine pathway, with prolactin and a full anterior pituitary panel. A craniopharyngioma can compress the optic chiasm and the hypothalamic-pituitary stalk, producing growth failure, delayed puberty, visual loss, and diabetes insipidus together, and the cost of delay is permanent visual and endocrine damage. [10] [5]
The second priority is to recognise the reversible functional causes and act on them immediately. An adolescent girl with low body mass, intense exercise, and amenorrhoea has functional hypothalamic suppression; the intervention is nutritional rehabilitation and reduction of training load, supported by a multidisciplinary eating-disorder team where anorexia nervosa is present, with bone-density assessment because the hypo-oestrogenic state is depleting her skeleton. Poorly controlled inflammatory bowel disease, cystic fibrosis, coeliac disease, and chronic kidney disease each need optimisation of the underlying condition — treating the disease restores the axis in a way that exogenous sex steroids cannot. Hypothyroidism and hyperprolactinaemia are treated directly with levothyroxine or dopamine agonists respectively. [11] [10]
The third priority is psychological support. The late-maturing adolescent — especially a boy who is the shortest and least developed in his peer group — carries a real burden of bullying, low self-esteem, social withdrawal, and sometimes depression. Acknowledge this explicitly, explain the diagnosis in plain language with the family, and frame the plan around a timeline. For the adolescent with constitutional delay and significant distress, a short course of testosterone both accelerates pubertal change and reassures the adolescent (and the family) that the body can respond — a powerful intervention in its own right. [12] [1]
Management — Definitive & Stepwise
Definitive management is tailored to the aetiology, and the Endo-ERN clinical practice guideline on pubertal induction and transition provides the framework. The guiding principle is to mimic normal puberty: introduce sex steroids at low dose and escalate gradually over 18 to 24 months so that secondary sexual characteristics develop at a physiological pace, final height is preserved, and psychosocial maturation keeps step with the physical changes. [9] [11]

For constitutional delay of growth and puberty, the management is reassurance and monitoring. Explain that the clock is intact and will start, provide a realistic timeline based on bone age, and review at six-monthly intervals for evidence of spontaneous progression. When the psychological burden is significant — typically in boys over 14 with marked short stature or peer difficulties — a short course of intramuscular testosterone (50 to 100 milligrams monthly for three to six months) induces the early changes of puberty, demonstrates that the body can respond, and provides a pubertal growth spurt that the constitutional-delay adolescent then continues on his own. The Stancampiano structured approach emphasises that testosterone in this context is a diagnostic and therapeutic trial, given at a dose and duration that does not advance bone age excessively or compromise final height. [12] [1]
For permanent congenital hypogonadotropic hypogonadism and the gonadal-failure group, pubertal induction is undertaken with escalating sex steroids. In boys, intramuscular or transdermal testosterone is started at a low dose (for example testosterone enantate 50 to 100 milligrams monthly intramuscularly, or a low-dose transdermal gel) and increased gradually over 18 to 24 months to a full adult replacement dose. In girls, oestrogen is introduced as low-dose transdermal oestradiol or oral ethinyloestradiol, started at a fraction of the adult dose and escalated over two to three years; a progestogen is added once breakthrough bleeding occurs or after adequate oestrogen priming, to establish regular cycles and protect the endometrium. The Endo-ERN guideline stresses gradual escalation to protect final height and psychosocial maturation, and to achieve physiological breast development in girls. [9] [11]
For the gonadal-failure syndromes, induction is followed by lifelong sex-steroid replacement and disease-specific surveillance. In Klinefelter syndrome, testosterone replacement addresses the androgen deficiency, preserves bone and muscle mass, and supports mood and energy; early semen analysis and fertility counselling matter because spermatogenesis is impaired but micro-dissection testicular sperm extraction can recover sperm in many men. In Turner syndrome, the Gravholt guideline coordinates oestrogen induction with growth-hormone therapy, cardiac surveillance (echocardiography and MRI for bicuspid valve, coarctation, and aortic dissection risk), renal imaging, audiology, and thyroid screening. Fertility options — for CHH via pulsatile GnRH or gonadotrophins (human chorionic gonadotropin plus follicle-stimulating hormone), and for gonadal failure via oocyte or sperm donation or cryopreservation — are part of long-term adult care. [7] [8]
T.I.M.E. \u2014 the four management decisions
Specific Subtypes & Scenarios
Constitutional delay of growth and puberty is the prototype and the commonest single diagnosis, accounting for the majority of boys and about a third of girls referred for delayed puberty. The presentation is the short, well, prepubertal adolescent with a delayed bone age and a family history of late puberty, and the management is reassurance with optional short-course testosterone for psychological distress. The prognosis for final adult height and full pubertal development is excellent, though final height may sit a few centimetres below the mid-parental target. The Sedlmeyer case series and the pedigree studies of Sedlmeyer-Hirschhorn and Wehkalampi established the familial aggregation and complex inheritance that underpin the diagnosis. [2] [3] [4]
Kallmann syndrome is the olfactory variant of congenital hypogonadotropic hypogonadism, defined by anosmia or hyposmia in association with absent or incomplete puberty due to deficient gonadotropin-releasing hormone. The embryological basis is the failed migration of GnRH neurons and olfactory neurons from the olfactory placode. Associated features include midline defects (cleft lip or palate), unilateral renal agenesis, synkinesia, and hearing loss. The inheritance is heterogeneous (X-linked ANOS1, autosomal dominant FGFR1, autosomal recessive and oligogenic forms). Management is pubertal induction followed by lifelong sex-steroid replacement, with fertility achieved through pulsatile GnRH or gonadotrophins. Stamou and Georgopoulos review the phenotype and genotype spectrum. [6] [5]
Klinefelter syndrome (47,XXY) is the commonest chromosomal cause of primary gonadal failure, with an incidence near one in 600 male births. The classic phenotype is tall stature, small firm testes, gynaecomastia, and infertility, but the presentation is variable and many cases escape diagnosis until adulthood. In adolescence it presents with delayed or incomplete puberty, small testes that do not progress, and high gonadotrophins with a low testosterone. The Groth clinical update covers the multisystem burden — learning and behavioural vulnerabilities, increased metabolic and cardiovascular risk, osteoporosis, and the fertility implications. Management is testosterone replacement from induction onward, with fertility counselling and semen cryopreservation where feasible. [7] [10]
Turner syndrome (45,X or mosaic) affects roughly one in 2500 girls and is the leading cause of hypergonadotropic hypogonadism in a short girl. The streak ovaries produce little oestrogen, so puberty does not start spontaneously and induction is required. Short stature is near-universal and is managed with growth hormone. The Gravholt clinical practice guideline sets out the comprehensive surveillance: cardiac (echocardiography and MRI for bicuspid aortic valve, coarctation, and aortic dissection risk), renal (imaging for collecting-system anomalies), audiological, thyroid (autoimmune hypothyroidism is common), and the timing of oestrogen induction coordinated with growth-hormone therapy to optimise final height. [8] [9]
Complications & Pitfalls
The complications divide into the skeletal, the psychological, and the disease-specific. The skeletal complication is reduced peak bone mass: the rapid mineral accrual of adolescence is driven by sex steroids, and every year of untreated hypogonadism forfeits bone that is never fully recovered. A bone-density assessment (DEXA) is part of the work-up in the permanent hypogonadism group, and timely pubertal induction is the intervention that protects the skeleton. In the female athlete triad and anorexia nervosa, the bone loss is compounded by low body weight and low insulin-like growth factor, and fracture risk is elevated into adulthood. [9] [11]
The psychological burden of delayed puberty is real and often underestimated. The late-maturing boy in particular faces bullying, social exclusion, low self-esteem, and sometimes depression, and the academic and social costs accumulate during the years of waiting. Acknowledging the distress, providing a clear timeline, and offering a short course of testosterone when the burden is significant are part of good care, not cosmetic indulgence. The disease-specific complications belong to the underlying syndrome: the cardiac and aortic risk of Turner (dissection is a lethal and preventable event), the metabolic and fertility burden of Klinefelter, and the visual and endocrine consequences of an untreated craniopharyngioma. [12] [8]
The chief cognitive trap is the false reassurance of constitutional delay. Because constitutional delay is so common, there is a tendency to label every well late-maturing boy and discharge him, missing the permanent CHH that will declare itself only when the expected puberty never comes. The anosmia test, the neonatal history (micropenis, cryptorchidism), the growth trajectory (normal-to-tall rather than short-for-age), and the family history (of anosmia and infertility, not just of late puberty) are the features that should lower the threshold for further investigation. A period of structured watchful waiting is safe, but only if there is a genuine plan to re-evaluate and escalate. [5] [6]
Prognosis & Disposition
The prognosis is determined by the aetiology. Constitutional delay of growth and puberty carries an excellent prognosis: spontaneous puberty occurs, final adult height is achieved (typically a few centimetres below the mid-parental target but within the normal range), bone density normalises once sex steroids flow, and fertility is normal. The Sedlmeyer and Wehkalampi data support reassurance as the cornerstone, with short-course testosterone reserved for the distressed adolescent. [2] [4]
Permanent congenital hypogonadotropic hypogonadism carries a good prognosis with appropriate induction and lifelong replacement: secondary sexual characteristics develop, bone mass is protected, and fertility is achievable with pulsatile GnRH or gonadotrophins. A minority of CHH patients show partial reversal of the axis over time, with endogenous sex-steroid production reappearing in adulthood, which is a reason to re-test the axis periodically rather than assume permanence. Primary gonadal failure (Klinefelter, Turner, acquired) carries a chronic prognosis managed by lifelong replacement and disease-specific surveillance, with fertility achievable through assisted reproduction in many cases. [6] [7] [8]
Disposition is shared, long-term, multidisciplinary care. A specialist paediatric endocrine service owns the diagnostic confirmation, the induction protocol, the growth-hormone coordination (in Turner), and the genetic and fertility counselling. The general paediatrician or family doctor owns coordination, growth and pubertal monitoring, immunisation, and the front-line management of intercurrent illness and psychosocial support. Every transition from paediatric to adult endocrine care is a high-risk point at which the replacement regimen, the surveillance plan, and the fertility counselling must be re-communicated. The Endo-ERN guideline frames transition as a structured process, not a single handover. [9] [1]
Special Populations
The same delayed puberty behaves differently across populations because access, recognition, and comorbidity are unevenly distributed. In remote and Indigenous communities, later presentation, longer retrieval times, and higher rates of chronic disease (which drive the functional group) mean that the window between onset and treatment is longer — so telehealth access to a paediatric endocrine service and clear referral thresholds are disproportionately important. In migrant, refugee and asylum-seeking families, consanguinity raises the pre-test probability of recessive CHH, chronic undernutrition drives functional suppression, and language barriers complicate the anosmia testing and the counselling — a trained interpreter is essential for every endocrine discussion. [5] [10]
In adolescents with chronic disease — inflammatory bowel disease, cystic fibrosis, coeliac disease, chronic kidney disease, and eating disorders — delayed puberty is often functional and tracks the disease activity, so the priority is optimisation of the underlying condition rather than endocrine intervention. Oncology survivors carry a dual risk of gonadotoxic primary failure and of hypothalamic-pituitary radiation effects, and need structured endocrine late-effects surveillance. In transgender and gender-diverse youth, the question of pubertal suppression with a gonadotropin-releasing hormone agonist arises in the context of gender-affirming care, and the delayed-puberty framework informs the monitoring of bone density and growth during suppression — a multidisciplinary gender service owns this pathway. [9] [11]
In adolescents with disability and neurodiversity, pubertal assessment is complicated by communication barriers and by syndromic associations (Prader-Willi with hypothalamic dysfunction, for example), and the timing of induction must balance physical development against the capacity for menstrual and sexual health management. Across all groups, the psychosocial impact of delayed puberty is amplified in the adolescent who is already vulnerable, and the plan must address the young person's body image, peer relationships, and mental health alongside the endocrine targets. [12] [1]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: the clinical epidemiology that defines the spectrum of causes, the genetic studies that explain the familial aggregation and the permanent central defects, and the consensus guidelines that standardise induction and surveillance. The Sedlmeyer-Palmert case series (2002) defined the relative frequency of causes in a referred population, showing the dominance of constitutional delay in boys. The pedigree studies of Sedlmeyer-Hirschhorn (2002) and Wehkalampi (2008) established the familial aggregation and complex inheritance of constitutional delay, reframing it as a genetic trait rather than a variant of normal. [2] [3] [4]
The Palmert-Dunkel NEJM clinical practice review (2012) is the canonical synthesis of the approach to delayed puberty and the reference most cited in fellowship answers. The Howard review of the genetic basis of delayed puberty (2019) covers the oligogenic architecture that connects constitutional delay and congenital hypogonadotropic hypogonadism. The Stamou-Georgopoulos review of Kallmann syndrome (2018) and the Groth update on Klinefelter syndrome (2013) provide the syndrome-specific depth, while the Gravholt Turner guidelines (2017) set out the comprehensive surveillance that defines modern Turner care. [1] [5] [6] [7]
The management evidence is consolidated in the Endo-ERN clinical practice guideline on pubertal induction and transition (Nordenström 2022), which standardises the gradual sex-steroid escalation protocol across the permanent hypogonadism group, and in the Stancampiano structured testosterone approach (2019), which addresses the specific question of testosterone use in adolescent boys. The Klein family-practice approach to disorders of puberty (2017) and the Butler delayed-puberty review (2020) provide the generalist framing. These guidelines are concordant across jurisdictions on the timing thresholds, the gonadotrophin-based classification, and the principle of gradual induction. [9] [12] [10] [11]
In Australia and New Zealand, delayed puberty is managed within the public and private paediatric endocrine services based at the major children's hospitals in each state and at Starship in New Zealand, with growth-hormone and sex-steroid therapies funded through the Pharmaceutical Benefits Scheme and Pharmac respectively where the criteria are met. Access from rural and remote communities is supported by telehealth endocrine clinics and aeromedical retrieval for the rare acute central lesion. Turner syndrome surveillance follows the Gravholt guideline framework, and the transition to adult endocrine care is coordinated through young-adult transition clinics. Indigenous and migrant families are supported by interpreter services and by Aboriginal health workers and cultural mentors, recognising that chronic disease and undernutrition drive a disproportionate share of the functional group in these populations. [8] [9]
Exam Pearls
A fellowship candidate answering on delayed puberty should land six anchor points and avoid three classic traps. The anchors are the sex-specific timing thresholds (no testes by 14 in boys, no breasts by 13 in girls), the gonadotrophin-based central-versus-gonadal split, the constitutional-delay triad of short stature, delayed bone age and family history, the anosmia of Kallmann syndrome, the stigmata of Klinefelter and Turner, and the principle of gradual pubertal induction over 18 to 24 months. The traps are failing to measure gonadotrophins, missing a craniopharyngioma behind a delayed puberty label, and treating functional suppression with sex steroids instead of addressing the cause. The candidate who can separate constitutional delay from permanent CHH at the bedside, name the bone-mass cost of untreated hypogonadism, and describe the Endo-ERN induction protocol passes the station. [1] [9]
References
- [1]Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med, 2012.PMID 22296078
- [2]Sedlmeyer IL, Palmert MR. Delayed puberty: analysis of a large case series from an academic center. J Clin Endocrinol Metab, 2002.PMID 11932291
- [3]Sedlmeyer IL, Hirschhorn JN, Palmert MR. Pedigree analysis of constitutional delay of growth and maturation: determination of familial aggregation and inheritance patterns. J Clin Endocrinol Metab, 2002.PMID 12466356
- [4]Wehkalampi K, Widén E, Laine T, Palotie A, Dunkel L. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. J Clin Endocrinol Metab, 2008.PMID 18160460
- [5]Howard SR. The Genetic Basis of Delayed Puberty. Front Endocrinol (Lausanne), 2019.PMID 31293522
- [6]Stamou MI, Georgopoulos NA. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism, 2018.PMID 29108899
- [7]Groth KA, Skakkebæk A, Høst C, Gravholt CH, Bojesen A. Clinical review: Klinefelter syndrome--a clinical update. J Clin Endocrinol Metab, 2013.PMID 23118429
- [8]Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol, 2017.PMID 28705803
- [9]Nordenström A, Ahmed SF, van den Akker E, et al. Pubertal induction and transition to adult sex hormone replacement in patients with congenital pituitary or gonadal reproductive hormone deficiency: an Endo-ERN clinical practice guideline. Eur J Endocrinol, 2022.PMID 35353710
- [10]Klein DA, Emerick JE, Sylvester JE, Vogt KS. Disorders of Puberty: An Approach to Diagnosis and Management. Am Fam Physician, 2017.PMID 29094880
- [11]Butler G, Purushothaman P. Delayed puberty. Minerva Pediatr, 2020.PMID 32748610
- [12]Stancampiano MR, Lucas-Herald AK, Russo G, Ahmed SF. Testosterone Therapy in Adolescent Boys: The Need for a Structured Approach. Horm Res Paediatr, 2019.PMID 31851967