Paeds · endocrinology-diabetes-and-growth
Diabetes insipidus and polyuria-polydipsia
Also known as Diabetes insipidus · Central diabetes insipidus · Nephrogenic diabetes insipidus · Arginine vasopressin deficiency · Arginine vasopressin resistance · AVP-D · AVP-R · Primary polydipsia · Polyuria-polydipsia syndrome · Copeptin · Desmopressin
Fellowship guide to diabetes insipidus and the polyuria-polydipsia syndrome in children: the vasopressin-water balance axis from hypothalamic osmoreceptor to renal aquaporin-2, the three-way split of central (AVP deficiency) versus nephrogenic (AVP resistance) versus primary polydipsia, the water-deprivation test and the copeptin revolution, and the management divide between desmopressin, thiazide-amiloride-indometacin, and fluid restriction.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The organising idea is the vasopressin-water balance axis. Hypothalamic osmoreceptors sense a rising plasma osmolality, the posterior pituitary releases arginine vasopressin, and vasopressin acts on the renal collecting duct to reabsorb water and concentrate the urine. Diabetes insipidus breaks this axis in one of two places: at the source, where no vasopressin is made (central), or at the kidney, where vasopressin cannot act (nephrogenic). Primary polydipsia is the mirror image — the axis is intact, but a flood of drunk water suppresses vasopressin and mimics the same dilute polyuria. Knowing which link is broken is the fastest route to the right test and the right drug. [1] [4]
This page covers the full breadth of the paediatric polyuria-polydipsia syndrome: the physiology of vasopressin and aquaporin-2, the three-way classification, congenital and acquired causes, the presentation from the failure-to-thrive infant to the child after brain surgery, the water-deprivation test, the copeptin tests that have largely replaced it, and the divided management. It cross-links to the SIADH leaf for the opposite disorder of water balance rather than repeating it here. [1] [2]
Overview & Definition
Diabetes insipidus is the passage of large volumes of inappropriately dilute urine because the kidney fails to concentrate it. In children, polyuria means a urine output above roughly 2 litres per square metre per day, or more than 40 to 50 millilitres per kilogram per day. The urine is hypotonic (osmolality typically below 300 milliosmoles per kilogram), and the child compensates by drinking — the polydipsia that gives the syndrome its name. [3] [5]
The word "insipidus" means tasteless, distinguishing this dilute urine from the sweet urine of diabetes mellitus, and the two diseases share nothing but the polyuria. This is the first fork in every assessment: a high glucose points to osmotic diuresis from diabetes mellitus, while a low urine osmolality with a normal glucose points to a water diuresis and the polyuria-polydipsia syndrome. [5] [10]
An international working group has renamed the two forms of diabetes insipidus to reduce the dangerous confusion with diabetes mellitus. Central diabetes insipidus is now arginine vasopressin deficiency (AVP-D), and nephrogenic diabetes insipidus is arginine vasopressin resistance (AVP-R). The older names remain in wide clinical use, so a fellowship candidate should recognise both and understand why the change was made. [2]
Classification
Once osmotic diuresis is excluded, the water diuresis divides into three diagnoses along one question: is vasopressin absent, ignored, or switched off? The figure below sets out that split and the causes under each branch. [1] [4]
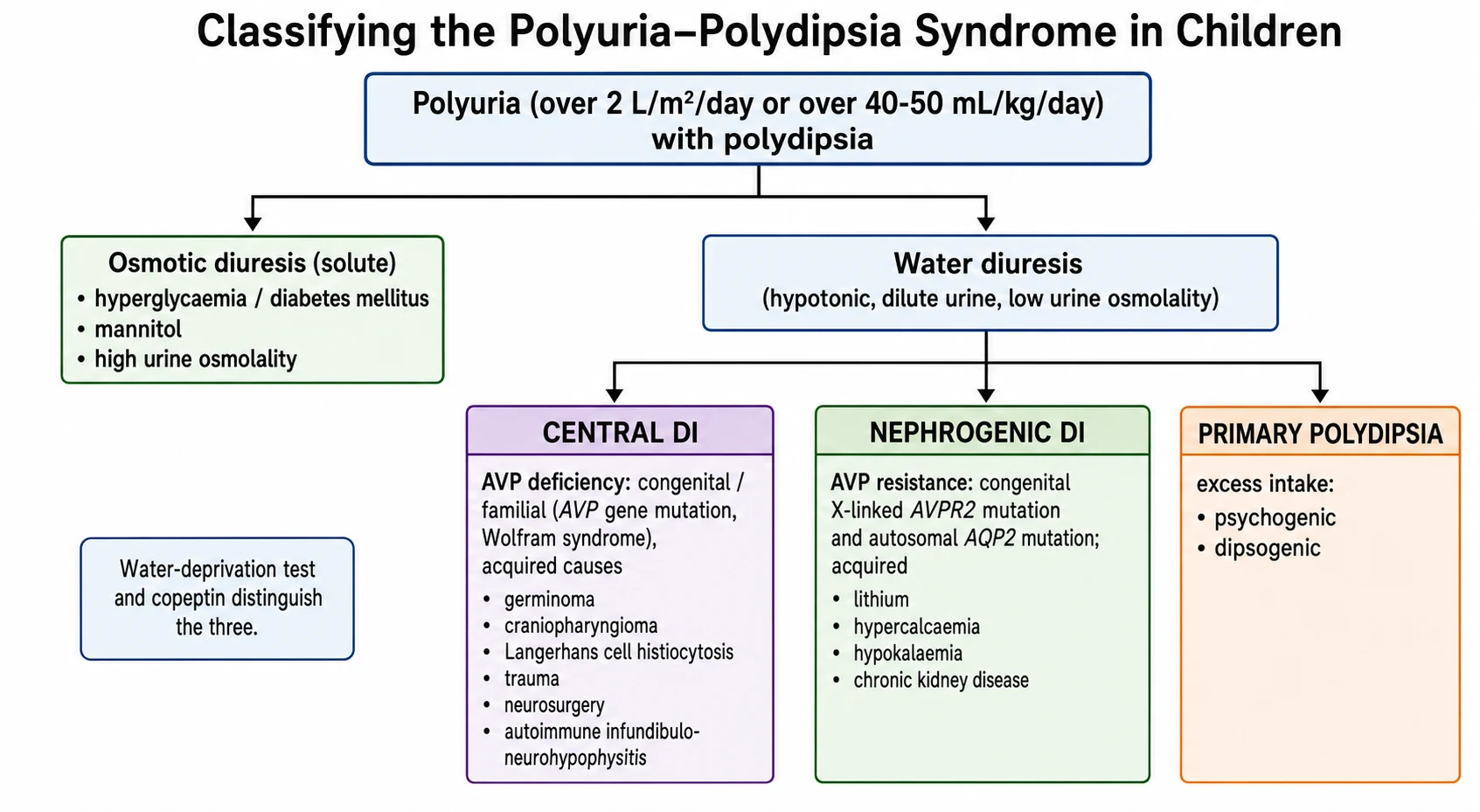
Central DI (AVP deficiency)
- Vasopressin not made or released — the break is at the source
- Congenital: AVP gene mutation, Wolfram (DIDMOAD), septo-optic dysplasia
- Acquired: germinoma, craniopharyngioma, Langerhans cell histiocytosis, trauma, neurosurgery, autoimmune
- Urine concentrates after desmopressin; copeptin is low
Nephrogenic DI (AVP resistance)
- Kidney cannot respond to vasopressin — the break is at the receptor or channel
- Congenital: X-linked AVPR2 (~90%), autosomal AQP2 (~10%)
- Acquired: lithium, hypercalcaemia, hypokalaemia, chronic kidney disease
- No response to desmopressin; copeptin is high
Primary polydipsia
- Excess water intake suppresses vasopressin — the axis itself is intact
- Psychogenic (behavioural or psychiatric) and dipsogenic (low thirst threshold)
- Chronic water load washes out the medullary gradient (partial concentrating defect)
- Urine concentrates with fluid restriction; desmopressin is dangerous here
Epidemiology & Risk Factors
Diabetes insipidus is uncommon in childhood, and the mix of causes shifts with age. In infancy the dominant concern is congenital nephrogenic disease, which presents in the first weeks of life and is easily mistaken for sepsis or feeding intolerance. In older children, acquired central disease from tumours, surgery, and infiltrative disease becomes the more common picture. [3] [4]
Central diabetes insipidus in children is most often acquired. Germ cell tumours (germinoma), craniopharyngioma, and Langerhans cell histiocytosis account for a large share, alongside traumatic brain injury and neurosurgery near the hypothalamic-pituitary region. A substantial minority are labelled idiopathic at first, and this group matters: some harbour an occult germinoma or Langerhans cell histiocytosis that only declares itself on later imaging, which is why surveillance is essential. [4] [11]
Congenital nephrogenic diabetes insipidus is a genetic disease of the collecting duct. About 90 per cent of inherited cases are X-linked, from mutations in the AVPR2 gene encoding the V2 receptor, so affected infants are usually boys; the remaining 10 per cent are autosomal, from mutations in the AQP2 gene encoding the aquaporin-2 water channel, and affect both sexes. Acquired nephrogenic disease has different risk factors: lithium therapy, hypercalcaemia, hypokalaemia, and chronic kidney disease. [9]
DIPS
Large volumes of hypotonic urine (osmolality below 300) with a normal glucose — a water diuresis, not osmotic
Primary polydipsia is excess intake; diabetes insipidus is an inability to conserve water (central or nephrogenic)
Acquired nephrogenic disease from hypercalcaemia, hypokalaemia, and lithium — reversible if caught
Idiopathic central diabetes insipidus needs surveillance MRI and tumour markers — germinoma and histiocytosis can appear late
Primary polydipsia occurs in children with behavioural or psychiatric drivers of drinking and in those with a low thirst threshold (dipsogenic). It is the hardest of the three to separate biochemically, because a long-standing water load washes out the medullary concentration gradient and blunts the kidney's ability to concentrate — so the water-deprivation test can look falsely like partial diabetes insipidus. [5] [6]
Pathophysiology
Water balance runs on a tight feedback loop between the brain and the kidney. When plasma osmolality rises by even one or two per cent, hypothalamic osmoreceptors trigger vasopressin release from the posterior pituitary and drive thirst. Vasopressin travels to the kidney, binds the V2 receptor on the collecting duct principal cell, and — through Gs, adenylyl cyclase, cyclic AMP and protein kinase A — moves aquaporin-2 water channels into the apical membrane. Water is reabsorbed down the osmotic gradient, urine concentrates, plasma osmolality falls, and vasopressin secretion switches off. [1] [9]
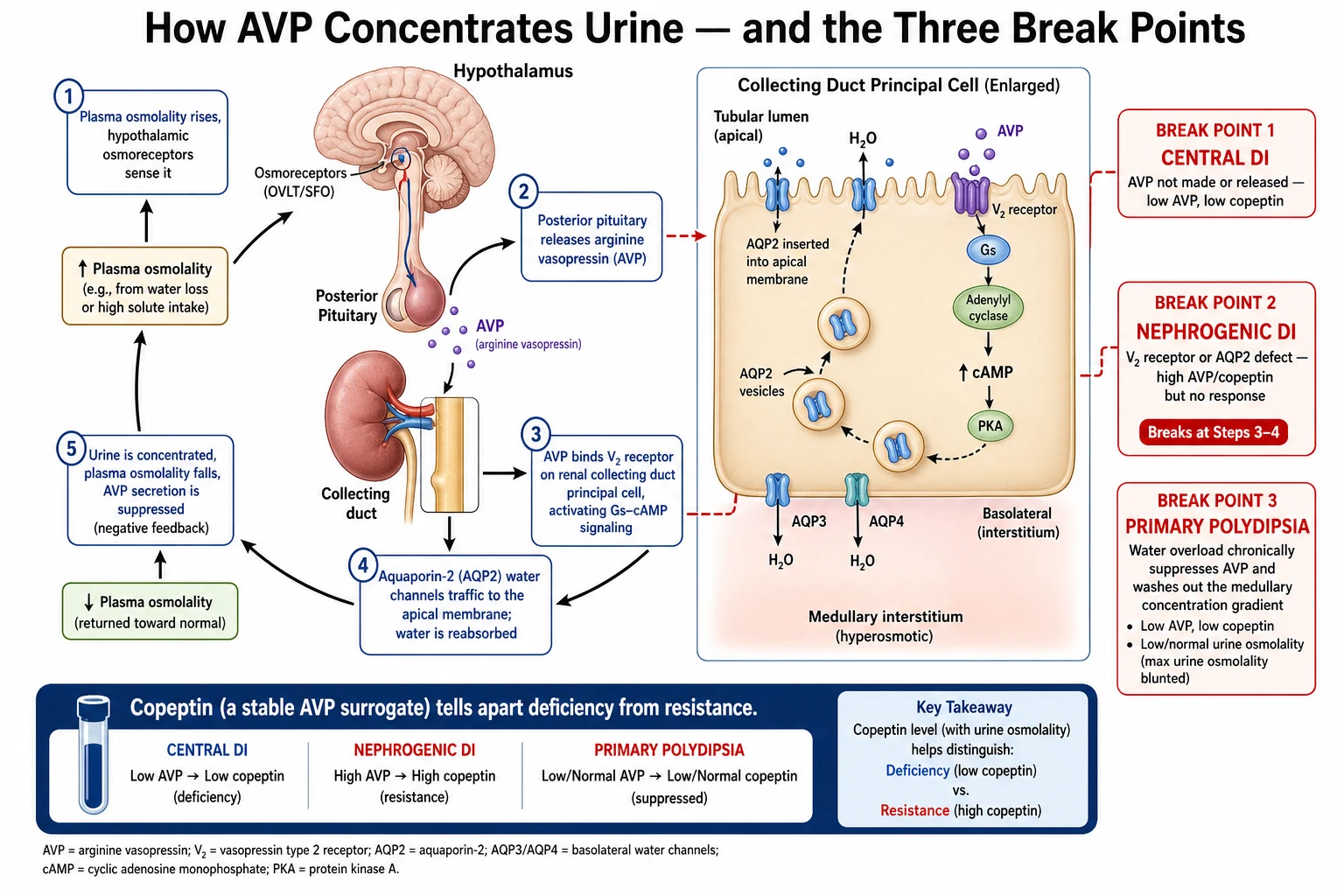
In central diabetes insipidus the break is at the source. Damage to the hypothalamic nuclei, the pituitary stalk, or the posterior pituitary means too little vasopressin is synthesised or released, so the kidney never receives the signal to concentrate. Because the magnocellular neurons that make vasopressin sit high in the hypothalamus, lesions of the stalk alone can still cause disease, and a lesion low in the stalk may spare enough neurons to leave only partial deficiency. [1] [4]
In nephrogenic diabetes insipidus the break is at the kidney. Vasopressin is made and released normally — indeed copeptin is high — but the collecting duct cannot respond. In the congenital forms a mutated V2 receptor cannot signal (AVPR2) or a mutated water channel cannot insert (AQP2). In the acquired forms, lithium enters the principal cell and downregulates aquaporin-2, while hypercalcaemia and hypokalaemia impair the concentrating machinery of the medulla. [9]
Primary polydipsia inverts the logic. The axis is structurally intact, but the child drinks so much that plasma osmolality falls, vasopressin is appropriately suppressed, and the urine is dilute. Over time the persistent water load washes solute out of the renal medulla, so the concentrating gradient is diminished and the kidney cannot fully concentrate even when challenged — the reason primary polydipsia can masquerade as partial diabetes insipidus. [5] [6]
Clinical Presentation
The cardinal complaint is drinking and passing large volumes of water around the clock. Older children describe unquenchable thirst, a preference for cold or iced water, nocturia, and secondary bedwetting in a previously dry child. The polyuria is constant, day and night, which helps separate it from the daytime frequency of a small bladder or a urinary tract infection. [4] [10]
Infants present very differently and far more dangerously. A baby cannot ask for water, so congenital nephrogenic diabetes insipidus shows itself as recurrent unexplained fevers, irritability, vomiting, constipation, poor feeding, and failure to thrive, often with episodes of hypernatraemic dehydration. These infants are frequently investigated for sepsis or gastrointestinal disease before the sodium and the paired osmolalities reveal the diagnosis. Repeated hypernatraemia in infancy also threatens neurodevelopment. [3] [9]
A specific and dangerous variant is adipsic diabetes insipidus, where the same hypothalamic damage that removes vasopressin also removes the sense of thirst. These children have lost both arms of osmotic defence: they cannot concentrate their urine and they do not feel the thirst that would otherwise protect them. They can climb to severe hypernatraemia without complaint, so they must be managed with scheduled fluids and daily weights rather than drinking to thirst. [11]
Differential Diagnosis
The differential of polyuria and polydipsia begins by separating a water diuresis from an osmotic diuresis. An osmotic diuresis carries a solute out with the water — glucose in diabetes mellitus, urea in the recovery phase after obstruction, or mannitol — and the urine osmolality is high. A water diuresis passes dilute urine with a low osmolality, and that is the gateway to the three diagnoses of diabetes insipidus and primary polydipsia. [5] [10]
Within the water diuresis, the discriminators are the serum sodium and osmolality, the response to desmopressin, and copeptin. A high or high-normal serum sodium with dilute urine favours true diabetes insipidus, because the child is losing free water faster than they can replace it. A low or low-normal serum sodium favours primary polydipsia, because the child is drinking ahead of their losses. Desmopressin then splits diabetes insipidus in two: central disease concentrates the urine, nephrogenic disease does not. [5] [10]
Points to central DI
- High or high-normal serum sodium with dilute urine
- Urine concentrates by more than 50% after desmopressin
- Low copeptin (baseline and after stimulation)
- MRI: loss of posterior pituitary bright spot, stalk lesion
Points to nephrogenic DI
- High serum sodium with dilute urine, often from infancy
- No concentration after desmopressin
- High copeptin (vasopressin is present but ignored)
- Family history, lithium, hypercalcaemia, or hypokalaemia
Points to primary polydipsia
- Low or low-normal serum sodium; large fluid intake
- Urine concentrates with supervised fluid restriction
- Copeptin response is adequate (axis intact)
- Behavioural or psychiatric context, or a low thirst threshold
A practical trap is that chronic primary polydipsia and partial central diabetes insipidus overlap on the classic water-deprivation test. Years of water loading blunt the medullary gradient, so a child with primary polydipsia may fail to concentrate fully and look like partial diabetes insipidus; conversely, partial central disease may concentrate somewhat and look like primary polydipsia. This overlap is exactly the problem that copeptin testing was designed to solve. [5] [7]
Clinical & Bedside Assessment
The assessment starts by confirming that the polyuria is real and quantifying it. Take a careful fluid diary — how much the child drinks and passes, whether it wakes them at night, and whether it is truly constant. Ask what they drink (a craving for iced water is characteristic of diabetes insipidus), and separate this from daytime urinary frequency, which is more often behavioural or infective. In a young child, weigh the nappies and chart the intake and output. [4] [10]
Take a targeted history for the cause. Ask about headache, visual change, and growth failure that might point to a hypothalamic-pituitary tumour; about head injury or neurosurgery; about drugs, especially lithium; and about a family history of infantile polyuria that suggests X-linked nephrogenic disease. In the infant, ask about recurrent fevers, vomiting, and poor weight gain, and review the growth chart, because faltering growth is a central clue. [3] [9]
Examine for the underlying disease and for the consequences of water loss. Check growth parameters and the visual fields for a suprasellar lesion, look for the skin rash and bony lesions of Langerhans cell histiocytosis, and assess hydration carefully in the acutely unwell infant. The examination rarely makes the diagnosis alone, but it steers the investigation and flags the child who needs urgent biochemistry rather than an elective work-up. [4] [11]
Investigations
The investigation moves in tiers: a first-line bundle that confirms a water diuresis and screens for the emergency, then a formal diagnostic test to separate the three causes, then imaging and tumour markers to find the underlying lesion. [5] [10]
Investigation bundle for the polyuria-polydipsia syndrome
Serum glucose — exclude diabetes mellitus and osmotic diuresis first
Paired serum and urine osmolality, with serum sodium — the core screen
Serum electrolytes, calcium, potassium, urea and creatinine — look for acquired nephrogenic causes
First-morning urine osmolality — a well-concentrated first-morning sample makes DI unlikely
Formal water-deprivation test, or a copeptin-based test (hypertonic saline or arginine stimulation)
Desmopressin challenge after deprivation — central DI concentrates, nephrogenic DI does not
Pituitary and hypothalamic MRI if central DI confirmed — bright spot, stalk, suprasellar lesion
Tumour markers (beta-hCG, alpha-fetoprotein) and surveillance imaging if idiopathic central DI
The water-deprivation test remains a standard tool where copeptin is unavailable. The child is deprived of fluid under close supervision while weight, urine osmolality, and serum osmolality or sodium are monitored. The test stops when the urine osmolality plateaus, the serum osmolality reaches a defined threshold, or the child loses more than 3 to 5 per cent of body weight. Desmopressin is then given: a urine osmolality that rises by more than about 50 per cent indicates central diabetes insipidus, little or no rise indicates nephrogenic disease, and a child who concentrates during deprivation has primary polydipsia. The test is laborious and can be unreliable, especially in partial forms. [5] [4]
Copeptin-based approach to diabetes insipidus (Fenske 2018)
N Engl J Med
Prospective study comparing hypertonic saline-stimulated copeptin against the indirect water-deprivation test for diagnosing the polyuria-polydipsia syndrome, using a reference diagnosis by an expert panel.
Key finding
Hypertonic saline-stimulated copeptin classified patients with substantially higher diagnostic accuracy than the water-deprivation test, particularly in separating primary polydipsia from partial central diabetes insipidus, which the deprivation test handled poorly.
Practice change
Stimulated copeptin measurement is now a preferred diagnostic strategy for the polyuria-polydipsia syndrome where the assay and safe sodium monitoring are available, reducing reliance on the cumbersome water-deprivation test.
Copeptin is the C-terminal fragment of the vasopressin precursor, released one-for-one with vasopressin but far more stable and easy to assay, so it works as a direct surrogate for vasopressin secretion. A high baseline copeptin, measured without prior fluid restriction, identifies nephrogenic diabetes insipidus, because vasopressin is present but ignored. To separate central diabetes insipidus from primary polydipsia the copeptin must be stimulated — either by hypertonic saline (raising the sodium to a target while monitoring closely) or, more simply and safely, by arginine infusion. A stimulated copeptin that stays low indicates central disease, while an adequate rise indicates primary polydipsia. [6] [7] [8]
Imaging follows a confirmed central diagnosis. Pituitary and hypothalamic MRI looks for loss of the normal posterior pituitary bright spot on T1, thickening or a lesion of the stalk, and a suprasellar mass. Because germinoma and Langerhans cell histiocytosis may not be visible at first, a child with idiopathic central diabetes insipidus needs beta-hCG and alpha-fetoprotein and repeated surveillance MRI over years, as the lesion can appear well after the water balance disorder. [4] [11]
Management — Resuscitation
The acute emergency in diabetes insipidus is hypernatraemic dehydration, and the two rules are to restore the circulation and then to correct the water deficit slowly. If the child is shocked, give an isotonic bolus to restore perfusion first. Once circulation is stable, replace the free-water deficit gradually, aiming to lower the serum sodium by no more than about 0.5 millimoles per litre per hour (around 10 to 12 millimoles per litre per day), because a sodium that falls too fast causes cerebral oedema and seizures. [3] [9]
Match the fluid to the ongoing losses. A child with diabetes insipidus keeps passing large volumes of dilute urine during resuscitation, so the maintenance and deficit fluid must account for those losses, and the sodium and fluid balance need frequent rechecking. In known central disease presenting acutely, desmopressin can be used, but it must be balanced against the fluid given so the sodium does not swing downward too quickly. [4] [9]
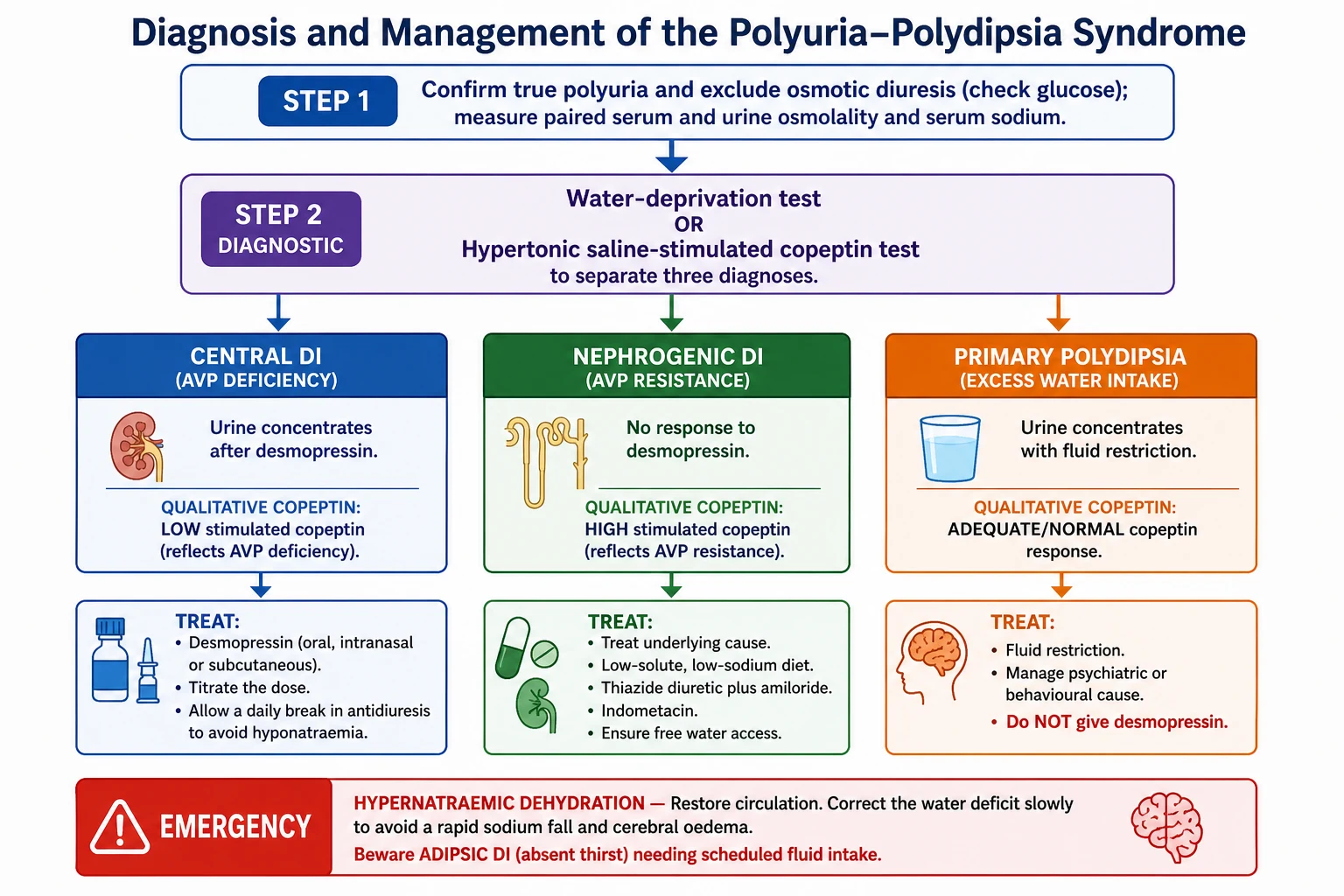
A distinct acute scenario is the post-neurosurgical triphasic response. After surgery near the pituitary, an initial diabetes insipidus phase (from stunned neurons) is followed around day 5 to 10 by a SIADH phase (as stored vasopressin is released from dying neurons), and then by permanent diabetes insipidus. If desmopressin is continued blindly into the SIADH phase, the child becomes dangerously hyponatraemic, so the sodium must be checked at least daily and the treatment adjusted to the phase. [4]
Management — Definitive & Stepwise
The definitive treatment follows the broken link. Central diabetes insipidus is treated with desmopressin, a synthetic vasopressin analogue that is a selective V2 agonist — it provides the missing antidiuresis without the vasopressor V1 effect of native vasopressin. It is given by the oral (tablet or melt), intranasal, or subcutaneous route, and the subcutaneous or intravenous route is far more potent than the intranasal one, so the routes are not interchangeable milligram for milligram. The dose is titrated to the lowest that controls the polyuria. [3] [4]
Desmopressin (DDAVP) — central diabetes insipidus
The single most important safety principle with desmopressin is to allow a daily break in antidiuresis. If the drug is dosed so that the child is antidiuretic around the clock and continues to drink, water is retained and the sodium falls, risking hyponatraemic seizures. Letting the polyuria break through briefly before each dose lets the child excrete a water load and protects against water intoxication. In neonates and young infants desmopressin is used very cautiously, and some are managed with careful feeding, fluid strategies, or thiazides instead, because their high obligate fluid intake makes hyponatraemia especially easy to provoke. [4] [3]
Nephrogenic diabetes insipidus cannot be fixed with desmopressin, so treatment reduces the water the kidney must handle and removes any reversible cause. Stop the offending drug (lithium) and correct hypercalcaemia or hypokalaemia. Provide a low-solute diet (restricting sodium and giving age-appropriate protein) to lower the obligate urine volume, and ensure unrestricted access to water. A thiazide diuretic paradoxically reduces urine output by inducing mild volume contraction and enhancing proximal water reabsorption; amiloride is added (especially in lithium-induced disease, where it blocks lithium entry through the sodium channel) and spares potassium; and indometacin or another prostaglandin inhibitor further potentiates concentration. [9]
Mechanism-based treatment of the three diagnoses
Central DI: start low-dose desmopressin, titrate to control polyuria, allow a daily antidiuretic break
Central DI: check the sodium regularly; use extra caution in neonates and the post-surgical triphasic phase
Nephrogenic DI: remove reversible causes — stop lithium, correct hypercalcaemia and hypokalaemia
Nephrogenic DI: low-solute low-sodium diet and free access to water to reduce obligate urine volume
Nephrogenic DI: thiazide diuretic, add amiloride (blocks lithium entry, spares potassium), add indometacin
Primary polydipsia: supervised fluid restriction and treat the behavioural or psychiatric driver
Primary polydipsia: do NOT give desmopressin — continued drinking on antidiuresis causes hyponatraemia
All: identify and treat the underlying cause, and arrange endocrine follow-up and, where relevant, tumour surveillance
Primary polydipsia is treated by reducing intake, not by antidiuresis. Supervised, graded fluid restriction lets the sodium normalise and the urine concentrate, and the behavioural or psychiatric driver is addressed with the relevant team. Desmopressin is contraindicated: a child who keeps drinking while their kidney is forced to retain water will develop hyponatraemia and can seize. Confirming the diagnosis before treatment is therefore not academic — it is the difference between safe and dangerous care. [10] [6]
Specific Subtypes & Scenarios
The scenarios below present acutely, confuse the team, or recur in exams. [4] [9]
Congenital nephrogenic diabetes insipidus presents in the first weeks to months of life, usually in a boy with an X-linked AVPR2 mutation. The infant has recurrent fevers, vomiting, irritability, constipation, poor feeding, and failure to thrive, punctuated by episodes of hypernatraemic dehydration. Early recognition matters because repeated hypernatraemia threatens neurodevelopment; management is a low-solute diet, free water, and the thiazide-amiloride-indometacin combination. [9] [3]
Idiopathic central diabetes insipidus is a diagnosis that must never be treated as final. A proportion of children first labelled idiopathic harbour a germinoma or Langerhans cell histiocytosis that becomes visible only later, so they need beta-hCG and alpha-fetoprotein and surveillance MRI of the pituitary and hypothalamus over years. Pituitary stalk thickening is a particular alarm for germinoma. [4] [11]
The post-neurosurgical triphasic response follows surgery near the pituitary: diabetes insipidus, then a SIADH phase around day 5 to 10, then permanent diabetes insipidus. The danger is running desmopressin blindly into the SIADH phase and causing hyponatraemia, so the sodium is checked daily and the treatment is matched to the current phase. [4]
Adipsic diabetes insipidus combines vasopressin deficiency with a lost sense of thirst, usually from extensive hypothalamic damage. These children cannot defend their water balance in either direction, so they are managed with a fixed fluid prescription, daily weights, and regular sodium checks rather than drinking to thirst. It carries the highest risk of severe hypernatraemia of any form. [11]
Complications & Pitfalls
The complications of diabetes insipidus come from the disease and from its treatment. The disease causes hypernatraemic dehydration, which in infants can cause seizures, cerebral injury, and — through repeated episodes — long-term neurodevelopmental harm. The treatment causes the opposite: desmopressin given without a daily antidiuretic break, or given at all in primary polydipsia, causes water retention, hyponatraemia, and hyponatraemic seizures. [3] [9]
Disease pitfalls
- Missing nephrogenic DI in a febrile, vomiting, faltering infant (mislabelled sepsis or gastroenteritis)
- Correcting hypernatraemia too fast — cerebral oedema and seizures
- Accepting an idiopathic central label without surveillance (missed germinoma or histiocytosis)
- Missing adipsic DI — no thirst means no protective drinking
Treatment pitfalls
- Desmopressin given to primary polydipsia — hyponatraemic seizures
- No daily antidiuretic break on desmopressin — water intoxication
- Running desmopressin into the post-surgical SIADH phase — hyponatraemia
- Treating routes of desmopressin as interchangeable (subcutaneous is far more potent)
The most dangerous acute error is correcting hypernatraemia too quickly. A serum sodium that falls faster than about 0.5 millimoles per litre per hour draws water into brain cells that have adapted to the high osmolality, causing cerebral oedema and seizures. The countermeasure is a planned, gradual correction with frequent sodium checks, replacing the free-water deficit over one to two days rather than hours. [9] [3]
The most dangerous chronic error is confusing primary polydipsia with diabetes insipidus and prescribing desmopressin. The child continues to drink, cannot excrete the retained water, and develops hyponatraemia. This is why the diagnostic separation — increasingly by stimulated copeptin — is a safety step, not a formality, and why fluid restriction rather than antidiuresis is the treatment for primary polydipsia. [6] [10]
Prognosis & Disposition
The prognosis depends on the cause and on the quality of long-term water management. Central diabetes insipidus is usually well controlled on desmopressin, and children live normally provided the dose is titrated safely and the sodium is monitored; the prognosis is then dominated by the underlying lesion, such as a germinoma or craniopharyngioma. Acquired nephrogenic disease from lithium, hypercalcaemia, or hypokalaemia often reverses when the cause is removed. [4] [9]
Congenital nephrogenic diabetes insipidus is lifelong, and its prognosis turns on preventing recurrent hypernatraemia. With early diagnosis, a low-solute diet, free water, and the thiazide-based regimen, growth and neurodevelopment can be protected, but repeated severe hypernatraemic episodes in infancy carry a real risk of lasting harm. Primary polydipsia has an excellent physical prognosis once the drinking is controlled, though the underlying behavioural or psychiatric driver needs ongoing attention. [9] [6]
Disposition is lifelong for the permanent forms. The child leaves on a clear water and medication plan, a named paediatric endocrinologist (and nephrologist for nephrogenic disease), a sick-day plan for illness and reduced intake, and — for adipsic disease — a fixed fluid prescription with daily weights. Children with idiopathic central disease carry a surveillance imaging plan, and all are set up for structured transition to adult services. [4] [11]
Special Populations
The young infant carries the highest acute risk. A baby with congenital nephrogenic diabetes insipidus cannot ask for water, presents with non-specific fevers and vomiting, and dehydrates rapidly, so the diagnosis is easily missed and the consequences of repeated hypernatraemia are serious. Desmopressin in central disease is used with special caution in this age group because the high obligate fluid intake makes hyponatraemia easy to provoke. [3] [9]
The child with adipsic diabetes insipidus has lost the thirst that normally rescues everyone else. They need a structured fluid regimen, daily weights, and regular sodium monitoring, because they will not drink in response to a rising osmolality and can become severely hypernatraemic silently. This is a chronic, complex, and technology-supported form of care. [11]
The child after craniopharyngioma or pituitary surgery often has combined pituitary deficits, and the water balance disorder sits alongside cortisol, thyroid, and growth hormone replacement. The adipsic variant is common after extensive hypothalamic surgery, and the triphasic response complicates the early post-operative period, so these children need coordinated endocrine and neurosurgical care. [4] [11]
The child with a developmental disability who cannot express thirst, or who is dependent on carers for fluids, is at particular risk from any disorder of water balance. Their care depends on carers understanding the fluid plan, recognising the early signs of dehydration, and having clear escalation advice, because the child cannot self-rescue by drinking. [9] [11]
Evidence, Guidelines & Regional Differences
The modern evidence base rests on the copeptin studies and the disease overview. The Christ-Crain Nature Reviews Disease Primers overview sets out the physiology and classification; the Timper multicentre study established baseline copeptin for identifying nephrogenic disease; the Fenske New England Journal of Medicine trial showed that hypertonic saline-stimulated copeptin outperforms the water-deprivation test; and the Winzeler Lancet study showed that arginine-stimulated copeptin is a simpler, safer stimulation. Together these have shifted diagnosis away from the cumbersome deprivation test where the assay is available. [1] [6] [7] [8]
An international working group has recommended renaming the disorders to arginine vasopressin deficiency and arginine vasopressin resistance, to end the confusion with diabetes mellitus that has caused real harm. This nomenclature is being adopted in the literature while the older terms persist clinically, so both should be recognised. The paediatric-specific guidance (Di Iorgi, Dabrowski) emphasises the tumour surveillance that separates children from adults, because idiopathic central disease in a child may be an occult germinoma or histiocytosis. [2] [4] [3]
Australia and Aotearoa New Zealand manage childhood diabetes insipidus through tertiary paediatric endocrinology, with nephrology co-management for nephrogenic disease. Desmopressin is available in oral, melt, intranasal, and parenteral forms, and stimulated copeptin testing is used in specialist centres where the assay is available, with the water-deprivation test still used elsewhere. Surveillance MRI and tumour markers for idiopathic central disease follow the international paediatric approach. [4] [7]
The genuine controversies are practical: how widely and how safely to deploy hypertonic saline-stimulated copeptin (which requires careful sodium monitoring) versus the gentler arginine stimulation, how long to continue surveillance imaging in idiopathic central disease, and how best to protect infants with nephrogenic disease from cumulative neurodevelopmental harm. A fellowship answer names the direction of travel — copeptin displacing the deprivation test — rather than pretending the field is fixed. [7] [8]
Exam Pearls
One-sentence answer: the approach to polyuria and polydipsia
A child with polyuria and polydipsia is assessed by first excluding osmotic diuresis with a glucose, then measuring paired serum and urine osmolality with a sodium, then separating central diabetes insipidus (low copeptin, concentrates with desmopressin), nephrogenic diabetes insipidus (high copeptin, no response to desmopressin), and primary polydipsia (adequate copeptin, concentrates with fluid restriction) — increasingly with stimulated copeptin rather than the water-deprivation test — and treating each by its mechanism: desmopressin, the thiazide-amiloride-indometacin regimen, and fluid restriction respectively.
Definition and classification
- Polyuria in children: over 40–50 mL/kg/day of dilute urine (osmolality below 300)
- Exclude osmotic diuresis (glucose) before calling it a water diuresis
- New names: central DI = AVP deficiency (AVP-D); nephrogenic DI = AVP resistance (AVP-R)
- Three-way split: central (no AVP), nephrogenic (AVP ignored), primary polydipsia (AVP suppressed)
Diagnosis
- Water-deprivation test: central DI concentrates by over 50% after desmopressin; nephrogenic does not
- High baseline copeptin = nephrogenic DI (AVP present but ignored)
- Stimulated (hypertonic saline or arginine) copeptin separates central DI (stays low) from primary polydipsia
- Copeptin outperforms the water-deprivation test, especially in partial and primary-polydipsia cases
Treatment by mechanism
- Central DI: desmopressin, titrated, with a daily antidiuretic break to avoid hyponatraemia
- Nephrogenic DI: remove cause, low-solute diet, thiazide + amiloride + indometacin, free water
- Primary polydipsia: fluid restriction and psychiatric/behavioural care — never desmopressin
- Hypernatraemia: correct slowly (sodium fall under ~0.5 mmol/L/hr) to avoid cerebral oedema
Paediatric traps
- Infant with fevers, vomiting, faltering growth and high sodium = nephrogenic DI
- Idiopathic central DI: surveillance MRI + beta-hCG/AFP (occult germinoma, histiocytosis)
- Post-pituitary-surgery triphasic response: DI → SIADH (day 5–10) → permanent DI
- Adipsic DI: no thirst — scheduled fluids and daily weights, not drinking to thirst
Frequently misremembered facts, stated correctly: in nephrogenic diabetes insipidus the vasopressin (and copeptin) is high, not low, because the hormone is present but the kidney ignores it. Thiazides reduce urine output in nephrogenic disease — a genuine paradox that works through mild volume contraction. Desmopressin is a selective V2 agonist with no pressor effect, and it is dangerous in primary polydipsia. And copeptin, not vasopressin itself, is the practical marker, because it is stable and easy to measure. [8] [9]
The one-diagram-that-answers-everything is the vasopressin-water axis: osmoreceptor senses, pituitary releases vasopressin, kidney reabsorbs water through aquaporin-2. Central disease breaks it at the source, nephrogenic disease breaks it at the kidney, and primary polydipsia suppresses it from above. Know where the break is, and the test (copeptin) and the treatment (desmopressin, thiazide, or fluid restriction) follow directly. [1] [7]
References
- [1]Christ-Crain M; Bichet DG; Fenske WK; et al Diabetes insipidus. Nat Rev Dis Primers, 2019.PMID 31395885
- [2]Working Group for Renaming Diabetes Insipidus; Arima H; Cheetham T; et al Changing the name of diabetes insipidus: a position statement of The Working Group for Renaming Diabetes Insipidus. Eur J Endocrinol, 2022.PMID 36239119
- [3]Dabrowski E; Kadakia R; Zimmerman D Diabetes insipidus in infants and children. Best Pract Res Clin Endocrinol Metab, 2016.PMID 27156767
- [4]Di Iorgi N; Napoli F; Allegri AE; et al Diabetes insipidus--diagnosis and management. Horm Res Paediatr, 2012.PMID 22433947
- [5]Fenske W; Allolio B Clinical review: Current state and future perspectives in the diagnosis of diabetes insipidus: a clinical review. J Clin Endocrinol Metab, 2012.PMID 22855338
- [6]Timper K; Fenske W; Kühn F; et al Diagnostic Accuracy of Copeptin in the Differential Diagnosis of the Polyuria-polydipsia Syndrome: A Prospective Multicenter Study. J Clin Endocrinol Metab, 2015.PMID 25768671
- [7]Fenske W; Refardt J; Chifu I; et al A Copeptin-Based Approach in the Diagnosis of Diabetes Insipidus. N Engl J Med, 2018.PMID 30067922
- [8]Winzeler B; Cesana-Nigro N; Refardt J; et al Arginine-stimulated copeptin measurements in the differential diagnosis of diabetes insipidus: a prospective diagnostic study. Lancet, 2019.PMID 31303316
- [9]Bockenhauer D; Bichet DG Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat Rev Nephrol, 2015.PMID 26077742
- [10]Robertson GL Diabetes insipidus: Differential diagnosis and management. Best Pract Res Clin Endocrinol Metab, 2016.PMID 27156759
- [11]Djermane A; Elmaleh M; Simon D; et al Central Diabetes Insipidus in Infancy With or Without Hypothalamic Adipsic Hypernatremia Syndrome: Early Identification and Outcome. J Clin Endocrinol Metab, 2016.PMID 26588450