Paeds · endocrinology-diabetes-and-growth
Disorders of sex development
Also known as disorders of sex development · differences of sex development · DSD · atypical genitalia · ambiguous genitalia · intersex disorders (deprecated)
A fellowship approach to disorders of sex development: recognise the atypical-genitalia infant, exclude a salt-wasting adrenal crisis before reaching for a label, classify using the 2006 Chicago framework built from karyotype rather than appearance, confirm with a karyotype and a 17-hydroxyprogesterone first, and deliver care through a multidisciplinary team that defers non-consent cosmetic surgery and protects lifelong psychosocial and gonadal health.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A baby is noted in the delivery room to have ambiguous genitalia — clitoromegaly and fused labia in an infant the midwife recorded as female, or a small phallus with severe hypospadias and impalpable gonads recorded as male. The fellowship task is to see that the appearance is only the surface, that beneath it may sit a metabolic emergency that closes in a fortnight, and that the name you eventually give the family is built from a karyotype and a hormone panel rather than from the external look. The wrong move is to assign a gender on the spot or to reach for a surgical fix before the picture is complete. [1] [4]
The four-move sequence for the atypical-genitalia infant
Overview & Definition
A newborn whose chromosomal, gonadal, or anatomical sex develops along an atypical path carries what the 2006 Chicago consensus named a disorder of sex development. The term was coined deliberately to retire the older vocabulary of 'intersex' and 'hermaphroditism', words that carried stigma and implied a person wedged between two sexes rather than a child with a specific, nameable, and treatable condition. The Chicago definition is deliberately broad — it captures any infant in whom the chromosomal sex, the gonadal sex, or the anatomical sex is unusual — because the point is to trigger a systematic work-up rather than to settle a label at the bedside. [1] [2]
The clinical importance of this definition is that it points the clinician to the right first question, which is never 'is this a boy or a girl' but 'what is the karyotype, and is there a metabolic emergency hiding behind this appearance'. Congenital adrenal hyperplasia caused by 21-hydroxylase deficiency is the single most common cause of atypical genitalia worldwide, and it is the one that kills through salt-wasting. Every other cause is a long, complex, multidisciplinary story, but the salt-wasting crisis is a two-week clock that starts ticking at birth. [3] [11]
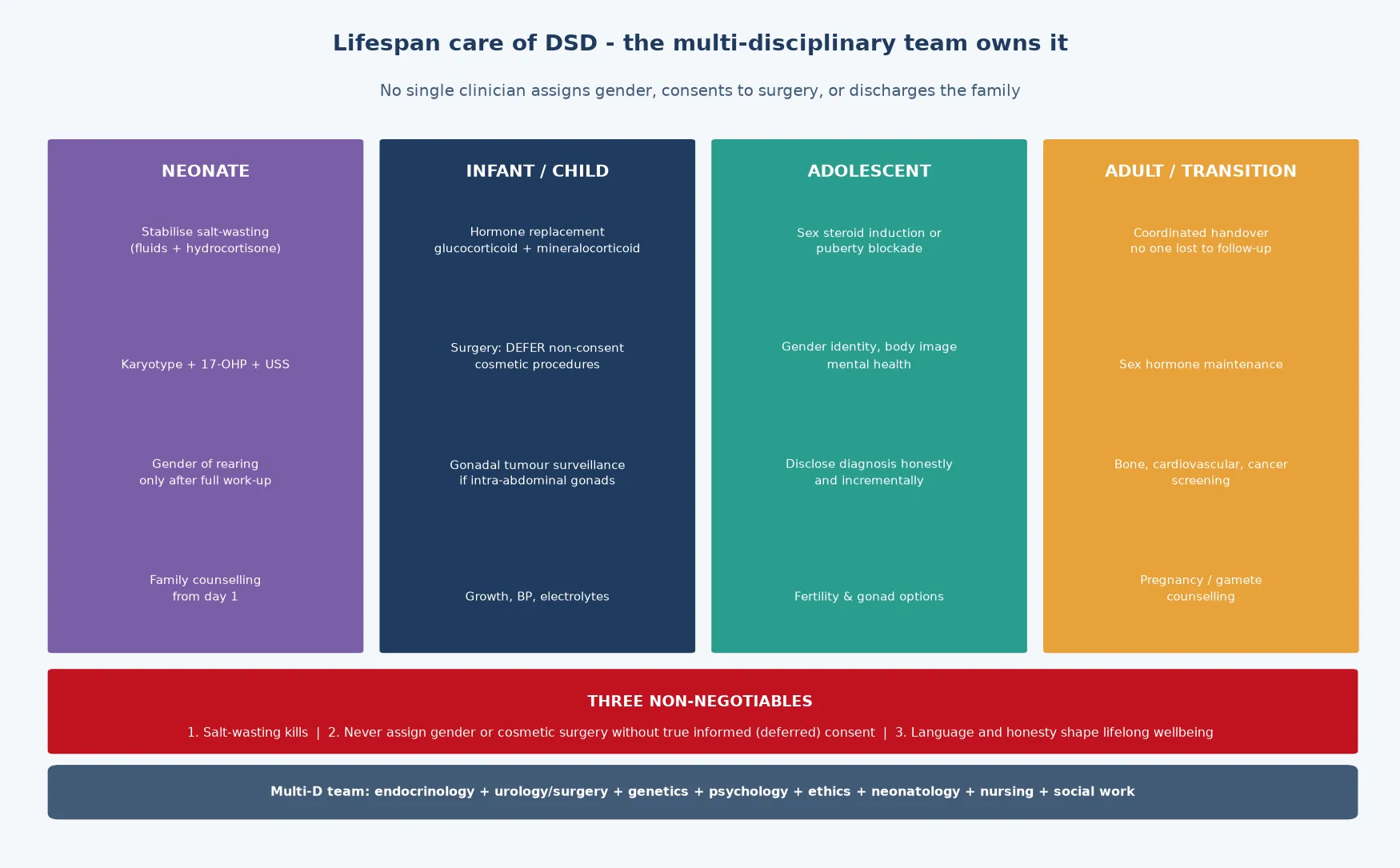
The condition sits at the intersection of endocrinology, genetics, urology, psychology, and ethics, which is why no single clinician owns it. The general paediatrician is the coordinator who stabilises the emergency, initiates the work-up, convenes the multidisciplinary team, counsels the family honestly, and stays with the child through puberty and into adult transition. The lifespan is long, the decisions are consequential, and the evidence on the best time to operate and disclose has shifted decisively toward deferral and honesty over the last two decades. [3] [7]
Classification
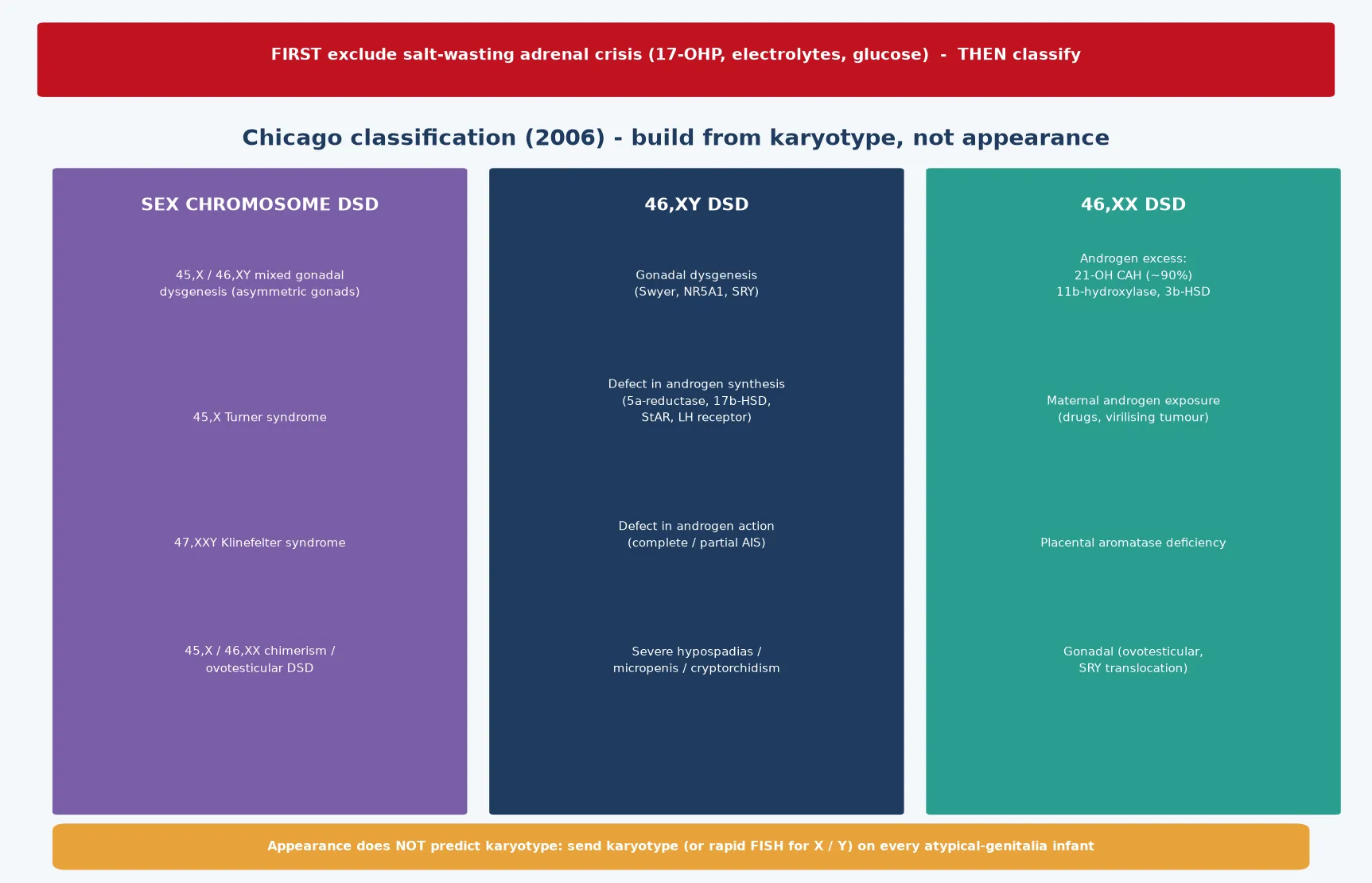
The Chicago consensus rebuilt the taxonomy around the karyotype, because the older labels pointed at appearances and at surgical options rather than at the underlying biology. The framework sorts every child into one of three bins: sex-chromosome DSD, where the chromosome complement itself is atypical; 46,XY DSD, where the chromosomes are male but development veered; and 46,XX DSD, where the chromosomes are female but androgen excess or gonadal anomaly masculinised the anatomy. Building from the karyotype forces the clinician to send the test that orients everything downstream, and it stops the bedside temptation to settle a gender from the external appearance. [1] [2]
Sex-chromosome DSD gathers the mosaic and aneuploid conditions, of which 45,X/46,XY mixed gonadal dysgenesis is the prototype — one side carries a streak gonad, the other a dysgenetic testis, and the phenotype runs a wide spectrum. Turner syndrome and Klinefelter syndrome also sit here, though they rarely present as neonatal atypical genitalia and instead declare themselves later in childhood or adolescence. Ovotesticular DSD, in which both ovarian and testicular tissue coexist, is grouped under this umbrella when the karyotype is mosaic, though it can also appear with a 46,XX complement. [6] [10]
The 46,XY bin holds the conditions in which a chromosomal male under-virilises, and the causes split into three mechanisms. Gonadal dysgenesis — the Swyer syndrome pattern, often from an SRY or NR5A1 defect — produces a streak gonad, normal Müllerian structures, and a female phenotype. Defects of androgen synthesis, such as 5α-reductase deficiency, 17β-hydroxysteroid dehydrogenase deficiency, and StAR protein mutations, leave a testis that cannot make enough testosterone to masculinise the external genitalia. Defects of androgen action, the androgen insensitivity syndromes, leave a target tissue that cannot respond to the testosterone that is present. [6] [7]
The 46,XX bin is dominated by androgen excess, and within that category one cause overwhelms every other. 21-hydroxylase deficiency congenital adrenal hyperplasia accounts for the great majority of 46,XX DSD, and it is the cause that carries the salt-wasting threat. The rarer 11β-hydroxylase and 3β-hydroxysteroid dehydrogenase deficiencies also virilise, but with different electrolyte and blood-pressure signatures. Maternal androgen exposure — from a virilising tumour or an androgenic drug — and placental aromatase deficiency masculinise a 46,XX fetus without a fetal adrenal cause, and the virilisation resolves the question when a careful maternal and drug history is taken. [6] [11]
Epidemiology & Risk Factors
Disorders of sex development are individually rare but collectively common enough that every general paediatrician will meet them. The most frequent single cause, 21-hydroxylase deficiency, occurs in roughly one in fourteen thousand to one in fifteen thousand live births in the classic form, though carrier frequencies are far higher and the non-classic form is among the commonest autosomal recessive disorders in some populations. The collective incidence of all atypical genitalia serious enough to warrant specialist evaluation sits around one in four to five thousand live births, which makes the atypical-genitalia infant a real event in any career. [6] [11]
The inheritance pattern matters for counselling and for the recurrence risk. The great majority of the enzyme defects in congenital adrenal hyperplasia are autosomal recessive, as are most of the androgen-synthesis defects, which means unaffected carrier parents face a one-in-four recurrence risk in each pregnancy and cascade testing is appropriate. Androgen insensitivity is X-linked recessive, carrying a different recurrence pattern and a maternal-carrier story. The gonadal dysgeneses are often sporadic but may follow an autosomal dominant or sex-linked pattern when a specific transcription-factor gene such as NR5A1 is involved. [7] [10]
Consanguinity intensifies the autosomal recessive burden, and certain founder mutations raise the incidence of specific enzyme defects in defined populations — Ashkenazi Jewish, Yupik Eskimo, and some Mediterranean communities carry higher 21-hydroxylase deficiency rates. These population facts matter in the antenatal clinic, where a known family history allows prenatal diagnosis and even antenatal dexamethasone to prevent virilisation in an affected 46,XX fetus, though the latter is now used selectively because of its own developmental uncertainties. [11] [3]
Newborn bloodspot screening for 21-hydroxylase deficiency is routine in Australia, New Zealand, and most developed systems, and it flags the salt-wasting infant before the crisis in many cases. The screen measures 17-hydroxyprogesterone, and its sensitivity depends on gestational age and birth weight, which is why premature and low-birth-weight infants generate false positives and require tiered interpretation. A normal screen does not exclude every form of congenital adrenal hyperplasia, so a clinically suspicious infant still needs a confirmatory work-up regardless of the bloodspot result. [11] [4]
Pathophysiology
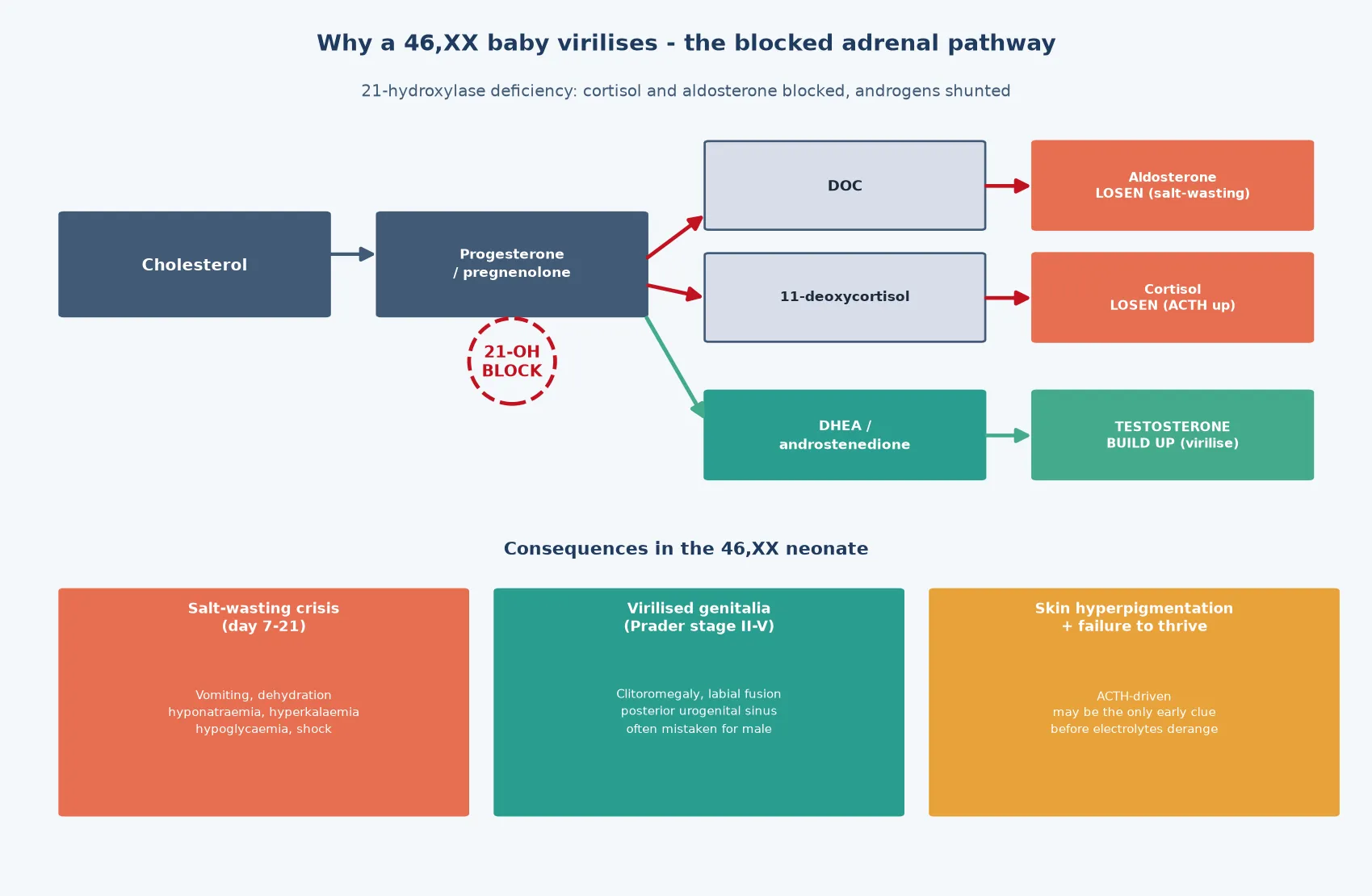
The molecular logic of the most dangerous DSD — 21-hydroxylase deficiency — sits in a single blocked enzyme step in the adrenal cortex. The enzyme 21-hydroxylase, encoded by CYP21A2, converts progesterone to 11-deoxycorticosterone on the mineralocorticoid pathway and 17-hydroxyprogesterone to 11-deoxycortisol on the glucocorticoid pathway. When the enzyme is absent, both mineralocorticoid and glucocorticoid production collapse, and the accumulating substrate is shunted down the only remaining route — the androgen pathway — where it is converted to testosterone. A 46,XX fetus bathed in androgen virilises, while the same infant loses the ability to retain sodium and to mount a cortisol response to stress. [11] [6]
The timing of that salt-wasting is what makes it lethal. The fetal adrenal is protected in utero by the placenta and the maternal supply, but after birth the infant must manage sodium and water alone, and the salt-losing form declares itself in the second or third week of life as vomiting, dehydration, hyponatraemia, hyperkalaemia, and hypoglycaemia. The crisis can progress to shock and death within hours of the first symptom, which is why the atypical-genitalia 46,XX infant is treated as a metabolic emergency before the karyotype even returns. The hyperpigmentation — visible at the scrotum, labia, and nipples — is the ACTH-driven clue that the pituitary is screaming at an unresponsive adrenal gland. [11] [2]
The 46,XY under-virilisation disorders follow a different logic, located downstream of the testis. A normal testis produces testosterone to masculinise the Wolffian ducts and external genitalia, and it produces anti-Müllerian hormone to regress the Müllerian structures. If the testis is dysgenetic — as in Swyer syndrome — it produces neither, and the fetus develops a female phenotype with a uterus and a streak gonad. If the testis makes testosterone but the enzyme 5α-reductase cannot convert it to dihydrotestosterone, the external genitalia under-virilise because the genital tubercle needs the more potent androgen. If the androgen receptor cannot respond at all, complete androgen insensitivity follows. [6] [7]
The gonadal malignancy risk that threads through these conditions is driven by the dysgenetic or intra-abdominal gonad rather than by the hormone defect itself. An intra-abdominal testis in partial androgen insensitivity or in gonadal dysgenesis carries a materially raised risk of germ-cell tumour, with the highest risk in the presence of a Y-chromosome-containing streak or dysgenetic gonad. The biology, mapped in the Hersmus and Looijenga work, explains why gonadal surveillance — by imaging, by biopsy, and sometimes by prophylactic removal after puberty — is built into every 46,XY DSD management plan. [8] [10]
Clinical Presentation
The atypical-genitalia neonate declares itself in the delivery room or on the routine newborn examination, and the pattern the clinician must recognise is the combination of features that does not fit either sex neatly. The virilised 46,XX infant shows clitoromegaly, posterior labial fusion, and a single urogenital opening, and may be mistaken for a male with bilateral cryptorchidism if the gonads are not palpated. The under-virilised 46,XY infant shows a small phallus, severe hypospadias, chordee, a bifid scrotum, and one or two impalpable gonads, and the degree of under-virilisation can range from nearly female to nearly male. [4] [5]
The single most reliable discriminator at the bedside is the presence or absence of palpable gonads. Gonads in the inguinal canal or scrotum are almost always testes, which means the infant is chromosomally or gonadally male, and the investigation turns toward under-virilisation. Bilaterally impalpable gonads in an apparent male raise the possibility of a virilised 46,XX infant with congenital adrenal hyperplasia — the must-not-miss diagnosis — because the 'gonads' recorded as undescended are in fact ovaries sitting safely inside the abdomen. This single examination finding redirects the entire differential and the entire emergency assessment. [4] [11]
The salt-wasting crisis presents not in the delivery room but in the second or third week of life, and it may be the first sign of congenital adrenal hyperplasia in an infant whose genitalia were recorded as normal or only mildly atypical. Vomiting, poor feeding, lethargy, weight loss, dehydration, and collapse draw the picture, and the biochemistry shows hyponatraemia, hyperkalaemia, hypoglycaemia, and a metabolic acidosis. A family history of a sibling who died in infancy of unexplained causes is a red flag for an undiagnosed salt-wasting disorder, and it should sharpen the threshold to send the electrolytes. [11] [2]
The later-presenting forms declare themselves outside the neonatal window. A pre-school or school-age child may present with premature pubic hair, clitoromegaly, or rapid growth from non-classic congenital adrenal hyperplasia. An adolescent may present with primary amenorrhoea — complete androgen insensitivity when the phenotype is female with normal breasts and absent uterus, or gonadal dysgenesis when the phenotype is female with no breast development and a high follicle-stimulating hormone. A virilising gonadal or adrenal tumour can present at any age with rapid and progressive masculinisation, and the speed of the change distinguishes it from the chronic enzyme defects. [6] [7]
Differential Diagnosis
The differential is structured by the karyotype, which is why the karyotype is sent before the differential is pursued. A bilaterally impalpable-gonad apparent male at the top of the list is congenital adrenal hyperplasia in a virilised 46,XX infant, and this must be excluded before any other cause is considered. Once a salt-wasting crisis is excluded or confirmed, the remaining differential sorts into the 46,XY under-virilisation causes — gonadal dysgenesis, androgen synthesis defects, and androgen insensitivity — and the rarer sex-chromosome mosaicisms. [4] [6]
Several syndromes produce overlapping phenotypes and must be separated. Smith-Lemli-Opitz syndrome, an autosomal recessive defect of cholesterol synthesis, produces 46,XY under-virilisation alongside intellectual disability, polydactyly, and a characteristic face, and it is excluded by a 7-dehydrocholesterol level. Denys-Drash and Frasier syndromes, both caused by WT1 mutations, produce gonadal dysgenesis with nephropathy and a high Wilms tumour or gonadoblastoma risk, which is why a renal ultrasound and blood pressure are part of the work-up of any 46,XY DSD with dysgenesis. [7] [10]
Transient and non-pathological causes must be excluded to avoid over-investigation. Maternal androgen exposure from a virilising luteoma, an androgenic drug such as a progestogen, or an aromatase-deficient placenta masculinises a 46,XX fetus and resolves without recurrence once the exposure ends, and a careful maternal drug and obstetric history closes the question. Isolated clitoral hood adhesions or labial synechiae in a well female infant are not DSD and do not warrant a karyotype. The discipline lies in sending the work-up for every genuinely atypical infant and in not medicalising normal variation. [5] [4]
Clinical & Bedside Assessment
The recognition move is to treat the atypical-genitalia infant as an emergency first and a diagnostic puzzle second. Examine the infant in a warm room with both gonads palpated, measure the phallus and the anus-to-genital distance, note the number and position of the urogenital openings, and record the Prader stage for a virilised 46,XX infant or the external masculinisation score for a 46,XY infant. The family is frightened and often ashamed, and the first conversation sets the tone for decades of care — the clinician names that this is a known condition, that it will be investigated systematically, and that no decision about gender or surgery will be made that day. [4] [5]
The history searches for the clues that narrow the cause. A family history of neonatal or infant death, of consanguinity, of amenorrhoea or infertility, or of a known DSD points to a genetic recurrence. A maternal drug history identifies androgenic exposures, and an obstetric history identifies maternal virilisation in pregnancy. The birth history records the gestation, the birth weight, and any perinatal stress, because prematurity affects the 17-hydroxyprogesterone reference range and because a sick infant raises the threshold for diagnostic certainty. [5] [6]
The examination extends beyond the genitalia. The clinician looks for hyperpigmentation of the scrotum, labia, and nipples that signals ACTH excess, for dysmorphic features that suggest a syndrome, for a cardiac murmur that may coexist with a chromosomal disorder, and for signs of dehydration or shock that indicate an evolving salt-wasting crisis. Blood pressure is measured, because hypertension in a virilised 46,XX infant points toward 11β-hydroxylase deficiency rather than the hypotensive 21-hydroxylase form. The whole child, not just the genitalia, is the assessment. [6] [11]
Investigations
The first-line panel is small and fast, and it orients every subsequent test. A karyotype or a rapid fluorescence in-situ hybridisation for the X and Y chromosomes establishes the chromosomal sex, and a 17-hydroxyprogesterone level identifies 21-hydroxylase deficiency. These sit alongside serum sodium, potassium, glucose, urea, and creatinine to detect the salt-wasting crisis, and a blood gas to detect acidosis. The result of the first panel — the karyotype and the electrolytes — redirects the second-line tests toward the right bin. [4] [11]
First-line laboratory panel for the atypical-genitalia infant
The second-line panel is built from the karyotype and the first results. In a 46,XX virilised infant with a raised 17-hydroxyprogesterone, the diagnosis is 21-hydroxylase deficiency and the work-up confirms the electrolytes and prepares for hydrocortisone and fludrocortisone. In a 46,XY under-virilised infant, the second-line tests measure testosterone, dihydrotestosterone, anti-Müllerian hormone, luteinising hormone, follicle-stimulating hormone, androstenedione, and dehydroepiandrosterone, and the ratios between them separate a synthesis defect from an action defect. A low testosterone with a raised luteinising hormone suggests a gonadal cause, a high testosterone with a low dihydrotestosterone suggests 5α-reductase deficiency, and a high testosterone with normal dihydrotestosterone suggests androgen insensitivity. [5] [6]
Imaging and genetic testing complete the picture. A pelvic and abdominal ultrasound identifies the uterus, the gonads, and the adrenal glands, and a genitogram defines the anatomy of the urogenital sinus and the vaginal cavity in the virilised infant. Genetic testing has moved from the targeted single-gene approach toward a multigene panel or chromosomal microarray, because many 46,XY DSD cases carry mutations in one of several known genes and the phenotype alone does not always predict which one. The goal of the work-up is a molecular diagnosis, because it informs the recurrence risk, the gonadal malignancy risk, and the long-term management. [7] [10]
Management — Resuscitation
The resuscitation of the salt-wasting crisis comes before the diagnosis, because the infant can die before the 17-hydroxyprogesterone returns. The presenting features are vomiting, dehydration, hyponatraemia, hyperkalaemia, hypoglycaemia, and shock, and the treatment is intravenous fluid resuscitation with normal saline, intravenous hydrocortisone at a stress dose, and intravenous dextrose to correct the hypoglycaemia. Hydrocortisone replaces the missing glucocorticoid and, at high doses, provides mineralocorticoid activity, and it is the single intervention that interrupts the spiral toward shock. Hyperkalaemia is managed by the hydrocortisone and the fluid, and rarely needs specific treatment once the cortisol is replaced. [11] [2]
The dose of hydrocortisone matters at the bedside. A salt-wasting infant is given an intravenous stress dose of hydrocortisone, typically a bolus followed by a continuous or divided maintenance replacement, and the sodium and potassium are monitored closely over the first hours because the potassium can transiently worsen before it corrects. Once the infant is stabilised, oral hydrocortisone replaces the glucocorticoid and oral fludrocortisone replaces the mineralocorticoid, and salt supplementation is added in infancy because the infant kidney is relatively insensitive to the mineralocorticoid. The family is taught the sick-day rules and the intramuscular hydrocortisone injection for emergencies before discharge. [11] [5]
The resuscitation of the family's understanding runs in parallel with the metabolic resuscitation, and it is no less urgent. The clinician names the condition honestly, avoids assigning a gender before the work-up is complete, and explains that the appearance will change with treatment in the case of congenital adrenal hyperplasia. The parents are warned that the gender of rearing is a decision the multidisciplinary team will make with them, that no surgery will happen that day, and that the child will be supported through childhood and adolescence. This conversation, done well, prevents the psychological harm that follows from secrecy and from premature surgical decisions. [3] [9]
Management — Definitive & Stepwise
The definitive management of congenital adrenal hyperplasia is lifelong hormone replacement, titrated to growth, electrolytes, blood pressure, and the renin and androgen markers. Glucocorticoid replacement with hydrocortisone suppresses the ACTH drive, and mineralocorticoid replacement with fludrocortisone manages the salt-wasting, with the doses adjusted through childhood as the child grows and the demands change. The aim is to suppress the excess androgen without over-treating, because over-treatment stunts growth and undertreatment allows virilisation and accelerated bone maturation. Monitoring is more frequent in infancy and around puberty, and the sick-day rules and the emergency injection are rehearsed with the family at every visit. [11] [5]
Hydrocortisone — glucocorticoid replacement in salt-wasting 21-hydroxylase CAH
The surgical question has shifted decisively over the last two decades, and a fellowship answer must reflect the shift. The 2006 consensus and the 2016 update moved toward deferring cosmetic genital surgery that is not medically necessary until the child can participate in the decision, because the long-term outcomes of early surgery — reduced sensation, dissatisfaction with appearance and function, and the psychological burden of a decision made without the person's consent — outweighed the convenience of operating in infancy. Medically necessary surgery, such as the repair of a urogenital sinus that causes recurrent infection, is performed on its own merits. The deferral principle applies most strongly to clitoral and vaginal surgery in 46,XX congenital adrenal hyperplasia, and the systematic review evidence on outcomes has hardened the case for waiting. [3] [12]
The gender-of-rearing decision is made by the multidisciplinary team with the family after the full work-up, and it is driven by the diagnosis, the predicted function, the likely gender identity, and the fertility potential. In 46,XX congenital adrenal hyperplasia, the gender of rearing is female because the chromosomes, the gonads, the internal structures, and the potential fertility are all female, and the virilisation reverses with treatment. In 46,XY partial androgen insensitivity, the decision is individualised to the degree of androgen responsiveness and the predicted gender identity. The decision is never made alone, it is never made under time pressure, and it is reversible if the evolving gender identity demands it. [3] [7]
Specific Subtypes & Scenarios
Congenital adrenal hyperplasia from 21-hydroxylase deficiency is the prototype and the most common cause, and it carries the salt-wasting threat. The virilised 46,XX infant is the classic neonatal presentation, and the under-virilised apparent male with impalpable gonads is the trap. Management is lifelong hydrocortisone and fludrocortisone with sick-day rules, and the surgical approach defers cosmetic procedures. The non-classic form presents later with premature pubic hair or rapid growth and is treated only if the symptoms warrant it. [11] [12]
Androgen insensitivity syndrome, complete and partial, is the other high-yield subtype. Complete androgen insensitivity presents at puberty with primary amenorrhoea, normal breast development from aromatised testosterone, absent or sparse pubic hair, and a blind-ending vagina with no uterus — the testis produces normal testosterone and anti-Müllerian hormone, but the androgen receptor is silent, so the phenotype is fully female. The intra-abdominal testis carries a malignancy risk that rises after puberty, and the gonad is removed after puberty with oestrogen replacement, or it is surveilled closely if the family and the team choose to retain it. Partial androgen insensitivity presents with variable atypical genitalia at birth, and the gender-of-rearing and surgical decisions are individualised. [7] [6]
Gonadal dysgenesis — the Swyer pattern in 46,XY and the mixed 45,X/46,XY form — carries a high gonadal malignancy risk from the dysgenetic or streak gonad, and the management centres on gonadal surveillance or prophylactic gonadectomy. The streak gonad in Swyer syndrome is removed after puberty because it carries a high gonadoblastoma and dysgerminoma risk, and the adolescent is induced through puberty with oestrogen then combined hormone replacement. The mixed gonadal dysgenesis child needs the dysgenetic gonad watched or removed according to its malignant potential, the karyotype, and the gender of rearing. [10] [8]
Complete androgen insensitivity — the lifespan arc
5α-reductase deficiency presents in a 46,XY infant with ambiguous genitalia, and it has a striking feature — virilisation at puberty. The testis produces testosterone normally, but the enzyme that converts it to the more potent dihydrotestosterone is deficient, so the external genitalia under-virilise before birth. At puberty the surge of testosterone is sufficient to masculinise through the androgen receptor, and many individuals raised female virilise and adopt a male gender identity. This subtype illustrates why the gender-of-rearing decision in 46,XY DSD is individualised and why deferring irreversible surgery protects the child. [6] [7]
Complications & Pitfalls
The salt-wasting crisis is the complication that kills, and missing it is the classic pitfall. An infant with vomiting and dehydration at two weeks of age is easily labelled gastroenteritis, and the electrolytes are not sent until the infant is in shock. The defence is to treat any atypical-genitalia infant — and any infant with a relevant family history — as a salt-wasting risk until the 17-hydroxyprogesterone and the electrolytes return, and to send them at the first symptom rather than after a trial of oral rehydration. The cost of a normal result is a blood test; the cost of a missed diagnosis is a death. [11] [2]
Over-treatment of congenital adrenal hyperplasia is a slow complication that stunts growth and suppresses the hypothalamic-pituitary axis, and it happens when the glucocorticoid dose is not reduced as the child stabilises. Under-treatment is the mirror pitfall, and it allows ongoing androgen excess, accelerated bone maturation, and final-height loss, with virilisation in the 46,XX child. The monitoring — renin, 17-hydroxyprogesterone, androstenedione, growth velocity, and bone age — is what keeps the dose in the narrow band between the two errors, and it is more frequent in infancy and around puberty. [11] [5]
The psychological complications of secrecy and of early non-consent surgery are the ones that surface in adolescence and adulthood, and the evidence on the harm of both has hardened over the last two decades. Adults who were operated on in infancy without their consent report higher rates of dissatisfaction with sexual function and appearance, and adults who were not told their diagnosis until late report higher rates of distrust and distress. The defence is honest, age-appropriate, incremental disclosure from childhood, and the deferral of cosmetic surgery until the person can consent. [3] [9]
Prognosis & Disposition
The prognosis in congenital adrenal hyperplasia is excellent with consistent replacement and sick-day management, and the limiting factors are adherence, intercurrent illness, and the quality of the transition to adult care. A child who takes the hydrocortisone and the fludrocortisone, who doubles the dose during illness, and who carries the emergency injection grows normally, enters puberty at the expected age, and can conceive with the appropriate reproductive technologies. The risks accumulate when the replacement is interrupted, when the monitoring lapses, and when the adolescent is lost to follow-up at the transition to adult care. [11] [3]
The prognosis in the 46,XY DSD subtypes depends on the diagnosis, the gonadal malignancy risk, and the gender identity trajectory. Complete androgen insensitivity carries a good quality-of-life prognosis with oestrogen replacement and psychological support, and the malignancy risk is managed by post-pubertal gonadectomy or surveillance. Gonadal dysgenesis carries a higher malignancy burden that demands structured surveillance or prophylactic removal. Across all subtypes, the psychosocial prognosis is shaped by the honesty of the disclosure, the timeliness of the support, and the absence of non-consent surgery, which is why the non-surgical and the communicative management are prognostic interventions in their own right. [7] [8]
The disposition is shared, structured, multidisciplinary care that runs from the neonatal unit to the adult endocrinology and psychology services. The general paediatrician coordinates the team — paediatric endocrinology, paediatric urology and surgery, clinical genetics, paediatric psychology, ethics, neonatology, nursing, and social work — and a named coordinator prevents the fragmentation that is the enemy of a long, complex plan. The transition to adult care is planned, documented, and rehearsed, because the loss of the adolescent to follow-up is the event that allows the bone-density, hormone, and malignancy surveillance to lapse. [3] [10]
Fertility and reproductive potential close the prognostic picture, and the counselling is built around them. A 46,XX child with congenital adrenal hyperplasia has normal ovaries and a uterus, and fertility is achievable with ovulation induction if needed. A 46,XY child with complete androgen insensitivity has no uterus and no fertility. The 46,XY subtypes with a functioning testis may have sperm retrieval options, and the gonadal dysgenesis subtypes may have fertility preserved if a streak gonad is removed before the germ cells are lost. The counselling is honest, specific to the diagnosis, and revisited at each life stage. [3] [7]
Special Populations
The atypical-genitalia infant and the DSD diagnosis interact with the family's social, cultural, and linguistic context, and the same management plan behaves differently across populations — access, cultural meaning, gender norms, and health literacy each shape the outcome. A plan that is clinically correct but delivered without cultural sensitivity or without an interpreter is no plan at all, and the fellowship answer recognises that the schedule is only as good as the family's ability to engage with it. [3] [5]
Indigenous children in Australia and New Zealand may carry a higher background burden of the conditions that present acutely, and reduced access to specialist endocrinology and genetics services in remote communities intensifies the need for early, structured referral. The salt-wasting crisis in a remote community is a retrieval as much as a diagnosis, and telehealth and outreach extend the multidisciplinary team into communities that a clinic-based model would miss. The cultural meaning of gender and of atypical genitality is handled with the family and, where appropriate, with the community, and the language is chosen with care. [3] [10]
Migrant, refugee, and asylum-seeking families may arrive with incomplete medical records, an uncertain family history, and consanguinity, and the diagnosis may carry a different cultural weight in the country of origin. A careful reconstruction of the family history, confirmation of the karyotype and the hormone panel, and an interpreter-mediated explanation of the diagnosis and the plan are the foundations. The written sick-day rules, the emergency injection, and the surveillance schedule are provided in the family's language, and the consent to any surgery is built on genuine understanding rather than on deference to authority. [5] [4]
Socioeconomic disadvantage shapes late presentation, access to the multidisciplinary team, and the feasibility of the sick-day rules and the emergency injection. The limiting step is often the cost of the medication, the transport to the clinic, and the time off work, rather than the medicine itself. Structuring the surveillance around a single coordinated visit, linking the family to a support organisation, and ensuring the medication is subsidised and available all improve engagement. The gender and sexual diversity of the child as they grow is protected from discrimination, and the team advocates for the child in the school and the community. [3] [9]
Evidence, Guidelines & Regional Differences
The evidence base rests on the 2006 Chicago consensus — the Hughes and Lee papers that unified the nomenclature and built the classification framework that every fellowship answer is structured around. The 2016 Lee update reframed the field around perceptions, approach, and care, and it moved the surgical and disclosure practices decisively toward deferral and honesty. The Ahmed UK guidance, revised in 2015, operationalises the first-line evaluation of the infant and the adolescent, and it is the practical structure of the work-up. [1] [2] [3] [5]
The congenital adrenal hyperplasia evidence is anchored by the 2018 Endocrine Society clinical practice guideline from Speiser and colleagues, which sets the replacement, the monitoring, and the surgical timing for 21-hydroxylase deficiency. The Almasri systematic review and meta-analysis on genital reconstructive surgery in congenital adrenal hyperplasia hardened the evidence on surgical outcomes and underpinned the move toward deferral. The Wisniewski 2019 Endocrine Reviews paper on the management of 46,XY DSD throughout life maps the lifespan arc, and the Hersmus work on the biology of germ-cell tumours anchors the gonadal malignancy surveillance. [11] [12] [7] [8]
Where the evidence is weak, a fellowship answer says so honestly. The optimal timing of surgery, the long-term gender-identity outcomes in the rarer subtypes, the role of antenatal dexamethasone to prevent virilisation, and the best approach to gonadal surveillance are areas of genuine and active debate, and the practices differ between centres and between countries. The move from 'disorders' toward 'differences' of sex development reflects a genuine, ongoing conversation about language, and the local terminology should be known. Naming the uncertainty is a mark of intellectual honesty that examiners reward. [3] [7]
In Australia and New Zealand, care follows the international consensus framework, with multidisciplinary DSD services in the major paediatric centres coordinating endocrinology, urology and surgery, genetics, psychology, ethics, neonatology, and nursing. Newborn bloodspot screening for 21-hydroxylase deficiency is routine across jurisdictions, though the threshold and the confirmatory pathway vary. The surgical deferral principle operates through state-based consent frameworks and through the specialist services, and the language has shifted toward 'differences' of sex development in many centres. Access to specialist services, genetic counselling, and adolescent psychology is uneven across rural and remote communities, which intensifies the need for early referral and for telehealth-supported surveillance. [3] [5]
Exam Pearls
A fellowship candidate answering on disorders of sex development should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for; the traps are where easy marks are lost. [1] [3]
Anchor one: exclude the salt-wasting crisis first. Any atypical-genitalia infant, and any bilaterally impalpable-gonad apparent male, is a salt-wasting risk until the 17-hydroxyprogesterone and the electrolytes return. The crisis declares itself at one to three weeks with vomiting, hyponatraemia, hyperkalaemia, and shock, and it kills before the label. [11]
Anchor two: build the diagnosis from the karyotype, not the appearance. Send a karyotype or a rapid FISH on every atypical-genitalia infant, because the appearance never predicts the chromosomal sex, and the karyotype orients every downstream test and decision. [2]
Anchor three: classify with the Chicago framework. The three-way classification — sex-chromosome DSD, 46,XY DSD, 46,XX DSD — built from the karyotype is the structure that earns the marks, and naming 21-hydroxylase deficiency as the most common cause and the salt-wasting threat anchors the epidemiology. [1] [6]
Anchor four: defer non-consent cosmetic surgery. The 2006 consensus and the 2016 update moved toward deferral, the outcome evidence hardened the case, and a fellowship answer names the principle without defensiveness. Medically necessary surgery is performed on its merits; the rest waits for the child's consent. [3] [12]
Anchor five: run the multidisciplinary team and the lifelong plan. Care runs from the neonatal unit to the adult endocrinology and psychology services, and the general paediatrician coordinates it. The surveillance covers hormones, growth, bones, gonads, fertility, and psychology, and the transition to adult care is planned and rehearsed. [3] [7]
The three traps to avoid are assigning a gender at the bedside before the work-up returns, performing cosmetic surgery in infancy without consent, and using the retired terms 'intersex' and 'hermaphrodite' with the family. The high-yield numbers a candidate holds are that 21-hydroxylase deficiency is the most common cause at roughly one in fifteen thousand live births, the salt-wasting crisis presents at one to three weeks, and the gonadal malignancy risk is highest in a dysgenetic gonad with a Y chromosome. Land the anchors, avoid the traps, and the answer reads as mature. [11] [8]
References
- [1]Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES Consensus Group, ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child, 2006.PMID 16624884
- [2]Lee PA, Houk CP, Ahmed SF, Hughes IA, International Consensus Conference on Intersex. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics, 2006.PMID 16882788
- [3]Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH, Mazur T, Meyer-Bahlburg HF, Mouriquand P, Quigley CA, Sandberg DE, Vilain E, Witchel S, International DSD Consortium. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm Res Paediatr, 2016.PMID 26820577
- [4]Ahmed SF, Rodie M. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol (Oxf), 2011.PMID 21521344
- [5]Ahmed SF, Achermann JC, Arlt W, Balen A, Conway G, Edwards Z, Elford S, Hughes IA, Izatt L, Krone N, Miles HL, O'Toole S, Perry L, Sanders C, Simmonds M, Watt A, Willis D. Society for Endocrinology UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development (Revised 2015). Clin Endocrinol (Oxf), 2016.PMID 26270788
- [6]Öçal G, Berberoğlu M, Saka N, Tükün A, Yağcı A, Akar N, Ruhi Hiözmenez H, Savaş Erdève S, Çetinkaya E, Açıkgöz Ç, Ensari A, Oktay G, Tarcan A, Adıyaman P, Semizoğlu F, Tokus M, Kandemir N, Akar HH, Topaloğlu AK, Kuloğlu Z, Hız S, Kılıç E, Kiper ÖC, Alikaşifoğlu A, Orhan D, Kale G. Current concepts in disorders of sexual development. J Clin Res Pediatr Endocrinol, 2011.PMID 21911322
- [7]Wisniewski AB, Batista RL, Costa EMF, Finlayson C, Sato N, Joy Lee P, McAndrew T, Rone MB, Defilippo RE, Hannema SE, Hughes IA, Baskin LS, Mendonca BB, Arlt W, Krstic-Zvelc D, Chitayat D, Cools M, D'Alberton F, Lee P, Looijenga LH, Reiner WG, Szarras-Czapnik M, Tadokoro-Cuccaro R, T'Sjoen G, Migeon CJ. Management of 46,XY Differences/Disorders of Sex Development (DSD) Throughout Life. Endocr Rev, 2019.PMID 31365064
- [8]Hersmus R, Stoop H, White SJ, Drop SLS, Oosterhuis JW, Incrocci L, Wolffenbuttel KP, Looijenga LHJ. The biology of germ cell tumors in disorders of sex development. Clin Genet, 2017.PMID 27716895
- [9]Crouch NS, Creighton SM. Minimal surgical intervention in the management of intersex conditions. J Pediatr Endocrinol Metab, 2004.PMID 15645692
- [10]McCann-Crosby B, Mansouri R, Dietrich KE, McCullough LB, Sutton VR, Austin EG, Roth DR, Karaviti LP, Gunter DF, Lee PA. State of the art review in gonadal dysgenesis: challenges in diagnosis and management. Int J Pediatr Endocrinol, 2014.PMID 24731683
- [11]Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HF, Miller WL, Murad MH, Oberfield SE, White PC. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2018.PMID 30272171
- [12]Almasri J, Zaiem F, Rodriguez Gutierrez R, Elraiyah T, Ahmad H, Al Nofal A, Ospina NS, Wang Z, Hay ID, Natt N, Murad MH. Genital Reconstructive Surgery in Females With Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab, 2018.PMID 30272250