Paeds · endocrinology-diabetes-and-growth
Growth hormone deficiency and excess
Also known as Growth hormone deficiency · GHD · Growth hormone excess · Pituitary gigantism · Paediatric acromegaly · Somatotropin deficiency · Recombinant human growth hormone therapy · rhGH therapy · Idiopathic short stature · X-linked acrogigantism
A fellowship approach to disordered growth hormone action: recognise the short child with growth hormone deficiency (short stature crossing centiles, delayed bone age, neonatal hypoglycaemia and midline defects) and the overgrowing child with growth hormone excess (accelerating growth velocity, headache, visual field defect), confirm with IGF-1 and a stimulation test or an oral glucose load, and treat with recombinant growth hormone titrated to IGF-1 for deficiency and transsphenoidal surgery with somatostatin analogue or pegvisomant for excess.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The mark goes to the candidate who reasons along three axes at once. The first is the child in front of you and the growth chart: a trajectory crossing centiles, a height far from the mid-parental target, or a growth velocity that is abnormal for age and sex. The second is the axis physiology: pulsatile growth hormone from the somatotrophs acting through hepatic and local IGF-1, with IGF-1 in turn feeding back to suppress growth hormone release. The third is the whole child: a growth hormone deficiency that is often part of multiple pituitary hormone deficiency and a midline malformation, and a growth hormone excess that is often a pituitary adenoma with mass effect on the optic chiasm and the rest of the pituitary. Treatment is lifelong and multidisciplinary, and the transition from paediatric to adult endocrine care is a high-risk moment that the candidate must own. [1] [10]
Overview & Definition
Growth hormone deficiency is the state in which the anterior pituitary somatotrophs fail to secrete enough growth hormone to sustain normal linear growth and metabolic homeostasis, producing short stature with a delayed bone age and, in early and severe forms, fasting hypoglycaemia and central adiposity. Growth hormone excess is the sustained over-secretion of growth hormone, almost always from a pituitary somatotropinoma, producing pathological linear growth while the growth plates are open — pituitary gigantism — and acral overgrowth with soft-tissue swelling and visceromegaly once the plates fuse, the paediatric counterpart of adult acromegaly. Both sit at the centre of the somatotrope axis, the neuroendocrine loop that runs from the hypothalamus through the pituitary and liver to the growth plate. [1] [10]
The axis is built around insulin-like growth factor 1. Growth hormone releasing hormone from hypothalamic neurones stimulates the somatotrophs to release growth hormone in pulsatile fashion, peaking at night and rising through puberty under the influence of sex steroids. Growth hormone acts on the liver to generate circulating (endocrine) IGF-1 and on cartilage and other tissues to generate local (paracrine) IGF-1, and IGF-1 then drives chondrocyte proliferation and linear growth at the growth plate while feeding back negatively on the hypothalamus and pituitary to restrain further growth hormone release. Insulin-like growth factor binding protein 3 is the principal carrier of IGF-1 in the circulation and is co-regulated by growth hormone, so a low IGF-1 with a low IGFBP-3 is the biochemical signature of growth hormone deficiency. [1] [3]
Classification
The clinically useful classification separates deficiency from excess, and then divides each by aetiology and by the integrity of the rest of the pituitary. Growth hormone deficiency is isolated (IGHD) or part of multiple pituitary hormone deficiency (MPHD), and it is congenital (genetic transcription-factor defects such as POU1F1, PROP1, HESX1; structural such as septo-optic dysplasia) or acquired (craniopharyngioma and other sellar or suprasellar tumours, cranial irradiation, traumatic brain injury, infiltrative disease). Growth hormone excess is classified by its genetic driver — sporadic somatotropinoma, AIP-mutant familial isolated pituitary adenoma, X-linked acrogigantism driven by GPR101 duplications, McCune-Albright syndrome, and multiple endocrine neoplasia type 1 — because the genotype dictates the age of onset, the aggressiveness, and the response to medical therapy. [1] [10] [12]
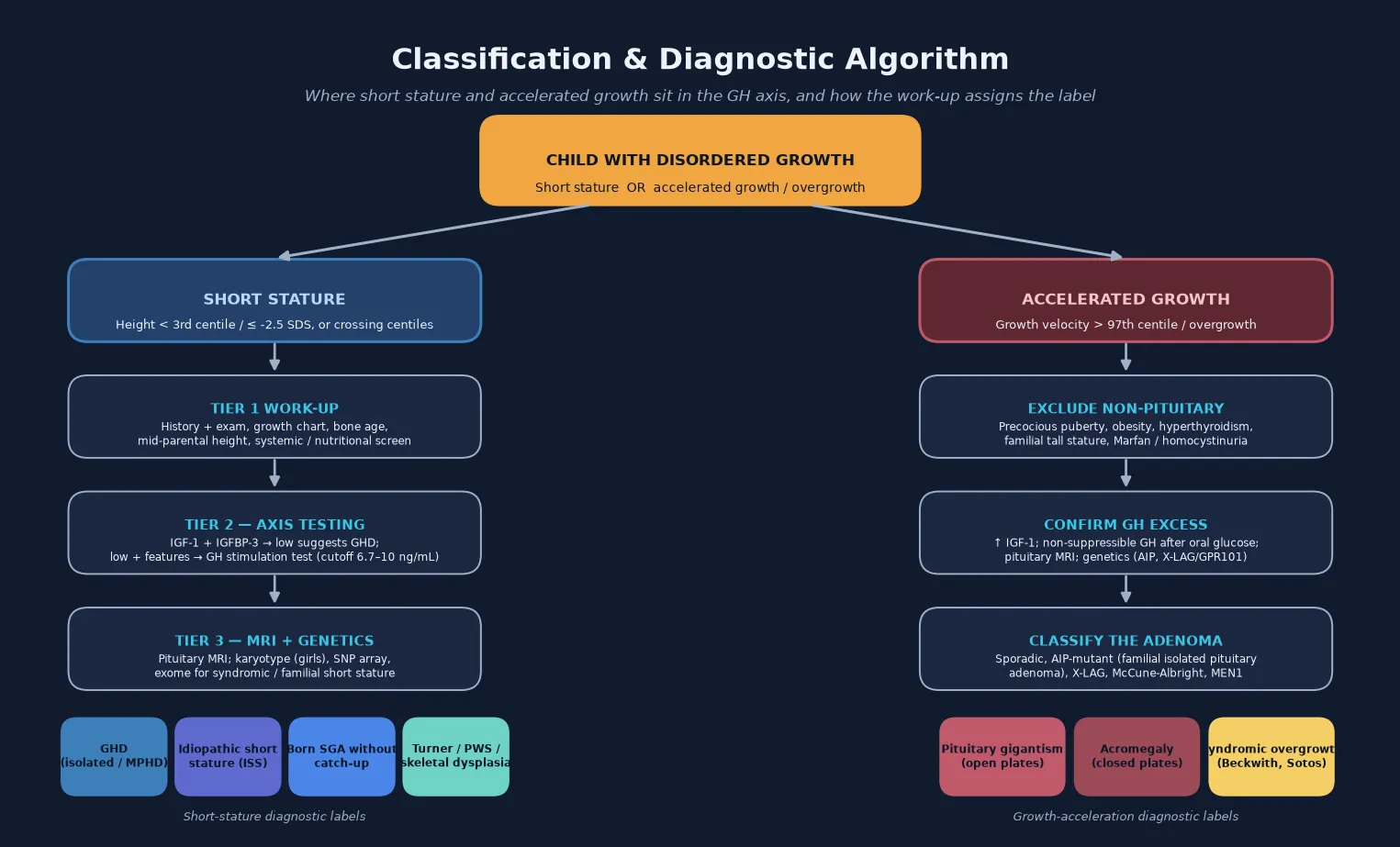
A parallel classification applies to the indications for recombinant growth hormone therapy, because growth hormone is prescribed for several distinct conditions in which the deficiency is relative rather than absolute. The licensed indications include proven growth hormone deficiency, Turner syndrome, children born small for gestational age who fail to show catch-up growth, Prader-Willi syndrome, chronic renal insufficiency, and idiopathic short stature. The dose, the expected response, and the safety profile differ across these groups, which is why the indication must be named precisely at the outset. [2] [6] [7]
Epidemiology & Risk Factors
Growth hormone deficiency affects roughly one in 3,500 to 10,000 children and accounts for a minority — perhaps 10 to 15 percent — of children presenting with short stature, the majority of whom have a familial, constitutional, or systemic cause. The male-to-female ratio in referred cohorts is skewed, partly reflecting ascertainment bias in boys, and the age at diagnosis clusters around the school-entry years when short stature becomes socially apparent — although congenital forms present in the neonatal period with hypoglycaemia and midline defects. Pituitary gigantism is rare by comparison, with somatotropinomas accounting for far less than five percent of all paediatric pituitary tumours, but the genetic subtypes (X-linked acrogigantism, AIP-mutant familial isolated pituitary adenoma, McCune-Albright) present at characteristic young ages and carry distinctive natural histories. [1] [10] [12]
The risk factors for growth hormone deficiency are the conditions that damage the hypothalamic-pituitary axis: suprasellar tumours (craniopharyngioma, germinoma), cranial irradiation (which causes progressive hypothalamic-pituitary failure, growth hormone deficiency often appearing first), traumatic brain injury, and the genetic and structural malformations. The risk factors for growth hormone excess are a family history of pituitary adenoma or hyperparathyroidism (suggesting MEN1 or AIP mutation), a known genetic syndrome, and a personal history of an earlier-onset tumour. Children born small for gestational age who do not demonstrate catch-up growth by the age of two to four years are a defined at-risk group for short stature that is licensed for growth hormone therapy. [3] [7]
Pathophysiology
The deficiency story begins with the failure of the somatotrophs to deliver enough growth hormone to maintain the IGF-1 that drives the growth plate. The chondrocytes of the proximal tibial and distal femoral growth plates proliferate in response to locally generated and circulating IGF-1, and when IGF-1 is low the cells proliferate more slowly, linear growth decelerates, and the bone age falls behind the chronological age — the hallmark that separates an endocrine growth failure from a constitutional one. At the same time, the metabolic actions of growth hormone falter: lipolysis falls and central adiposity accumulates, and the gluconeogenic and counter-regulatory actions weaken, so that a growth-hormone-deficient neonate fasts into hypoglycaemia because the axis that should sustain glucose during the overnight interval is absent. [1] [3]
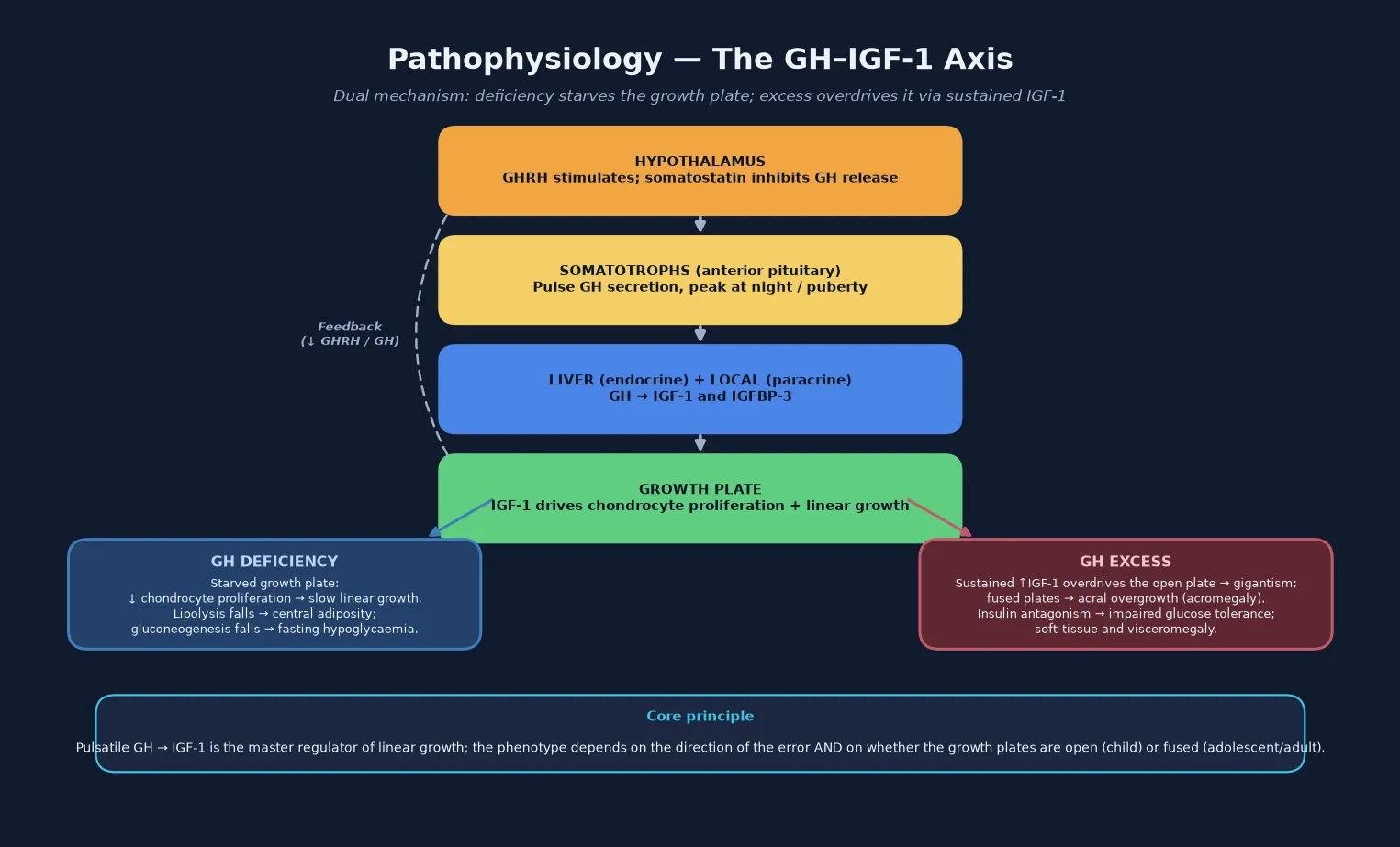
The excess story is the opposite end of the same axis. A somatotropinoma secretes growth hormone autonomously and continuously rather than in the normal nocturnal pulses, so IGF-1 is driven up and the growth plate is overstimulated — producing accelerated linear growth (gigantism) while the cartilaginous plates are still open. Because the tumour also produces the metabolic actions of growth hormone, lipolysis rises, soft tissue proliferates, and insulin signalling is antagonised, producing impaired glucose tolerance and occasionally overt diabetes. Once the growth plates fuse under the influence of sex steroids at the end of puberty, further growth hormone excess can no longer increase stature but instead drives acral overgrowth — enlargement of the hands, feet, jaw and skull, with visceromegaly, the syndrome then called acromegaly. A subtype worth knowing is X-linked acrogigantism, in which duplication of the GPR101 gene drives an early-childhood, often mixed somatotroph-lactotroph macroadenoma with extremely high growth hormone and prolactin, presenting in the first years of life. [10] [12]
The growth plate itself is the final common pathway, and its state — open or fused — is what decides whether growth hormone excess produces gigantism or acromegaly. This is why bone age assessment matters at both ends of the axis: a delayed bone age is consistent with deficiency, and the assessment of remaining growth potential in an overgrowing child determines whether the window for intervention on stature is still open. The dual mechanism — growth plate proliferation plus the metabolic actions of growth hormone on glucose, lipids and soft tissue — explains why treatment must normalise the IGF-1 and not merely the growth rate, because uncontrolled excess carries cardiovascular and metabolic risk that accumulates over decades. [10] [12]
Clinical Presentation
The presentation of growth hormone deficiency is dominated by the growth trajectory. The parent or the clinician notices that the child is the shortest in the class, that clothes are not being outgrown, and that the child looks younger than peers. The growth chart shows a height below the third centile, a height distant from the mid-parental target, and — most importantly — a growth velocity that is subnormal for age, typically below the 25th centile for bone age over a six-to-twelve-month interval. The child has a cherubic, immature face, increased central adiposity with preserved weight, a high-pitched voice, and delayed dentition; the bone age is delayed, often by two years or more. [1] [3]
The neonatal and congenital presentation is distinct and dangerous. A growth-hormone-deficient neonate may present with persistent hypoglycaemia (growth hormone is a key counter-regulatory hormone), a small penis in a phenotypic male (the so-called micro-penis of congenital hypogonadotrophic hypogonadism or panhypopituitarism), prolonged jaundice, or a midline defect — cleft lip or palate, a single central maxillary incisor, or septo-optic dysplasia with nystagmus and visual impairment. Recurrent hypoglycaemic seizures in this group cause preventable brain injury, and any neonate with persistent hypoglycaemia and a midline malformation or micropenis must have the pituitary axis checked. Congenital hypopituitarism with multiple hormone deficiencies can also present with adrenal crisis at the time of an intercurrent illness, because adrenocorticotrophic hormone deficiency is often part of the same developmental defect. [3]
The presentation of growth hormone excess is the mirror image: growth that is too fast. The parent notices rapid outgrowth of shoes and clothes, an increasing height that crosses centiles upward, and increasingly large hands and feet. Examination finds a growth velocity above the 97th centile for age and sex, prognathism and dental malocclusion, coarse facial features, oily skin and acne, sweating, headache, and — if the adenoma compresses the chiasm — a bitemporal hemianopia that is often missed until formal visual field testing is performed. Girls may present with menstrual irregularity or galactorrhoea (the tumour may co-secrete prolactin or stalk effect may elevate prolactin), and either sex may show impaired glucose tolerance. The diagnosis is frequently delayed because rapid growth in childhood is initially attributed to a normal variant of tall stature, and the candidate who checks the growth velocity and the IGF-1 early earns the mark. [10] [11]
Differential Diagnosis
The differential of short stature is broad, and growth hormone deficiency is only one part of it. The first task is to separate normal variants — familial short stature and constitutional delay of growth and puberty — from pathological short stature, and the growth velocity, the bone age, and the mid-parental height do most of this work. Familial short stature shows a child growing parallel to the centiles at a velocity appropriate for bone age, with a bone age concordant with the chronological age and parental heights that match. Constitutional delay shows a delayed bone age, a late pubertal timing often with a family history, and a growth velocity that is normal for bone age — and the child reaches a normal final adult height, just later than peers. [1] [3]
The pathological causes divide into systemic, endocrine, genetic and skeletal. Systemic disease — undiagnosed coeliac disease, inflammatory bowel disease, chronic kidney disease, chronic anaemia, cyanotic or congestive cardiac disease, and undernutrition — must be excluded with a careful history, examination, and a first-line screen (full blood count, coeliac serology, renal function, CRP). Endocrine causes include growth hormone deficiency, hypothyroidism, Cushing syndrome, and the rare glucocorticoid excess. Genetic and syndromic causes include Turner syndrome (every short girl must have a karyotype considered), Noonan syndrome, Prader-Willi syndrome, and SHOX deficiency. Skeletal dysplasias produce disproportionate short stature that the candidate detects with upper-to-lower segment and arm-span measurements. The principle is that the growth chart, the body proportions, the systemic screen, the bone age, and the IGF-1 together direct the work-up rather than a blanket endocrine panel on every short child. [1] [2]
The differential of accelerated growth is narrower but equally important to work through, because the first task is to exclude the benign causes before labelling a tumour. Familial tall stature shows tall parents, a child growing parallel to the centiles, and a normal growth velocity for the family. Precocious puberty shows early sexual development with an accelerated bone age that will compromise final height. Obesity can mildly accelerate growth in early childhood. Hyperthyroidism produces a growth spurt with other systemic features. Syndromic overgrowth — Sotos, Beckwith-Wiedemann, Marfan, homocystinuria — produces overgrowth with recognisable physical stigmata. Only after these are excluded does pathological growth hormone excess move to the top, and the high IGF-1 and the non-suppressible growth hormone after a glucose load confirm it. [10] [12]
Clinical & Bedside Assessment
The bedside assessment begins and ends with the growth chart. Measure the child standing (or lying supine for an infant) using a stadiometer, plot the height against population references, calculate the growth velocity from a previous measurement at least six months earlier, and measure and plot the mid-parental target height with its target range. Examine the body proportions (upper-to-lower segment ratio, arm span against height) to detect disproportion, look for dysmorphic features suggesting a syndrome, and assess the pubertal stage. In the short child, screen the systems for chronic disease and check for the stigmata of Turner syndrome in any girl. In the overgrowing child, examine the visual fields, the facial and acral features, the skin for café-au-lait patches of McCune-Albright, and the glucose. [1] [10]
The history captures the perinatal events (birth weight and gestation to identify the small-for-gestational-age child, neonatal hypoglycaemia, micropenis, jaundice), the feeding and nutritional pattern, the intercurrent illnesses and their effect on growth, the pubertal timing of the parents, and a three-generation family history of stature, puberty, and endocrine or pituitary disease. Ask specifically about headache, visual change, polyuria and polydipsia (diabetes insipidus in a midline tumour), and about symptoms of other pituitary deficiencies — fatigue, cold intolerance, hypoglycaemia, failure to thrive — or of excess — sweating, snoring, menstrual change. The examination is completed with a careful fundus and visual field assessment, because a bitemporal hemianopia from chiasmal compression may be the only clue to a pituitary macroadenoma. [3] [10]
The bedside assessment converts directly into the investigation plan. A low IGF-1 in the right clinical context, or a subnormal growth velocity, triggers the stimulation test and the pituitary imaging. A high IGF-1 in an overgrowing child triggers the oral glucose suppression test and the magnetic resonance imaging. The candidate who links the growth-chart abnormality to the next best investigation — rather than ordering a blanket panel on every short child — demonstrates the reasoning the exam rewards. [1] [10]
Investigations
The first-tier test for growth hormone deficiency is the insulin-like growth factor 1 and IGFBP-3, drawn in a non-fasting or standardised state and interpreted against age- and sex-specific reference ranges. A genuinely low IGF-1 in a well-nourished child with subnormal growth velocity raises the probability of growth hormone deficiency; a normal IGF-1 makes clinically significant deficiency less likely but does not exclude it, because IGF-1 is also influenced by nutrition and systemic illness. IGFBP-3 adds specificity in younger children, in whom IGF-1 is less discriminating. The supporting tests define the rest of the axis and exclude mimics: thyroid function (hypothyroidism suppresses growth hormone action and IGF-1), a systemic screen (coeliac serology, renal function, full blood count), a karyotype in any short girl, and a bone age radiograph. [1] [3]
Why the growth hormone stimulation test is needed — and why a single low IGF-1 is not enough
Growth hormone is secreted in pulses, so a random growth hormone level is uninterpretable. The diagnosis of growth hormone deficiency therefore rests on the peak growth hormone response to a pharmacological stimulus — clonidine, arginine, glucagon, insulin tolerance, or the newer and safer glucagon and arginine tests — with a cutoff historically at 10 ng/mL and now increasingly at 6.7 ng/mL using modern assays. The Guzzetti study demonstrated that the lower cutoff of 6.7 ng/mL improves specificity without sacrificing sensitivity in children with organic disease, and most contemporary guidelines accept a peak below 6.7 to 10 ng/mL as consistent with deficiency, with the cutoff and the number of tests (usually two, but one in a clearly deficient child with a defined structural lesion) set by local protocol. [5] [1]
The stimulation test is performed under supervision because the agents have side effects — clonidine causes hypotension and drowsiness, arginine causes nausea, insulin causes hypoglycaemia (and requires careful monitoring), and glucagon causes nausea and late hyperglycaemia. Growth hormone is sampled at intervals after the stimulus, and the peak is read against the cutoff. Two abnormal tests are generally required for a biochemical diagnosis of isolated growth hormone deficiency in a child with no structural lesion, although a single convincing test may suffice when the pre-test probability is high. Once the biochemical diagnosis is made, pituitary magnetic resonance imaging defines the anatomy — a small or ectopic posterior pituitary and a thin or absent stalk in congenital hypopituitarism, or a mass in an acquired lesion — and a full pituitary axis panel (TSH, free T4, cortisol, prolactin, LH, FSH, oestradiol or testosterone) excludes accompanying deficiencies. [3] [10]
For growth hormone excess, the first-tier test is a raised IGF-1 against age- and sex-matched references, confirmed by failure of growth hormone to suppress below 1 ng/mL after a 75-gram oral glucose load — the pathognomonic finding of autonomous somatotroph secretion, because glucose normally suppresses growth hormone. Pituitary magnetic resonance imaging with gadolinium defines the adenoma and its relationship to the optic chiasm, and formal visual field testing documents chiasmal compression. Genetic testing for AIP, GPR101 duplication (X-linked acrogigantism), MEN1, and the GNAS mutation of McCune-Albright is now part of the work-up of a paediatric somatotropinoma, because the genotype guides surveillance of the family and the choice of medical therapy. [10] [11] [12]
Management — Resuscitation
Growth hormone disorders rarely require resuscitation in the way an adrenal crisis or diabetic ketoacidosis does, but two situations demand immediate action. The first is the neonate with congenital hypopituitarism presenting with hypoglycaemia. The glucose must be corrected with intravenous dextrose, the airway and breathing secured, and a critical sample drawn if possible. Concurrent cortisol deficiency must be assumed and treated with hydrocortisone until adrenal insufficiency is excluded, because an ACTH-deficient neonate can present in shock during illness. The second is the macroadenoma with acute visual loss (pituitary apoplexy or rapidly progressive chiasmal compression), which is a neurosurgical and endocrine emergency requiring urgent imaging, corticosteroid cover, and ophthalmology and neurosurgery involvement. [3] [10]
In the overgrowing child with a confirmed somatotropinoma, the immediate priorities before definitive treatment are to protect the chiasm, to replace any deficient pituitary hormones (especially cortisol and thyroid), and to control the metabolic consequences — glucose intolerance may need treatment, and the systemic effects of excess growth hormone (hypertension, cardiac dysfunction) must be assessed. The child with a very large or aggressive tumour, particularly the AIP-mutant or X-linked acrogigantism subtypes, may need medical therapy to reduce tumour size and growth hormone secretion before surgery is safe, and a paediatric endocrinologist and neurosurgeon should be involved from the outset. [10] [11]
Management — Definitive & Stepwise
The definitive management of growth hormone deficiency is recombinant human growth hormone, given as a once-daily subcutaneous injection at bedtime to mimic the physiological nocturnal pulse. The standard starting dose is 0.045 to 0.050 milligrams per kilogram per day, titrated to maintain the IGF-1 in the upper half of the age- and sex-normal range and to optimise growth velocity while avoiding supraphysiological exposure. Higher doses are licensed for Turner syndrome (0.050 to 0.067 mg/kg/day) and for small-for-gestational-age children without catch-up growth (0.035 to 0.067 mg/kg/day), reflecting the relative growth hormone resistance in these conditions. Therapy is continued until near-final height is reached, and then the patient is re-tested to determine whether adult growth hormone deficiency persists and warrants continuation. [1] [2]
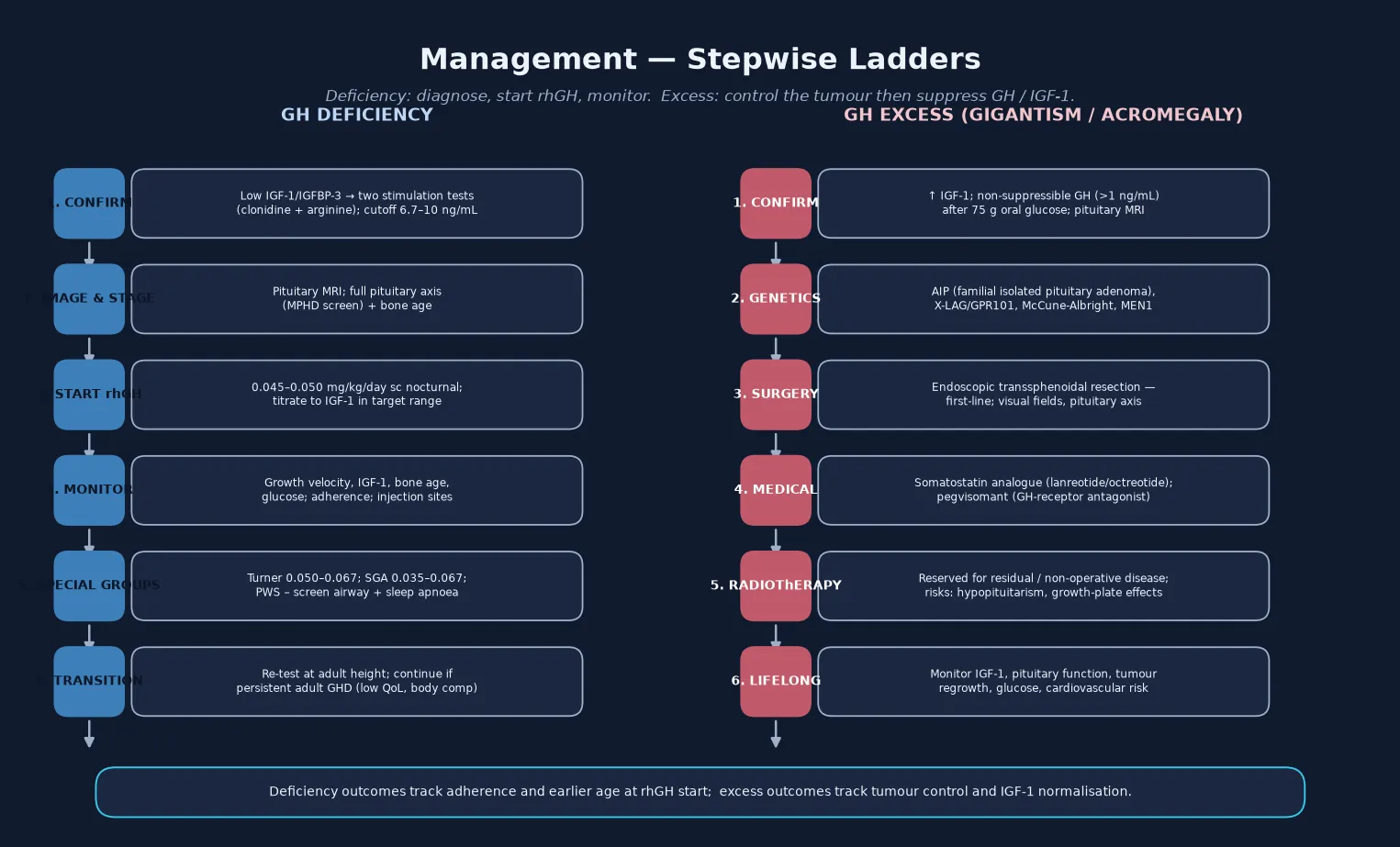
Monitoring is the discipline that makes recombinant growth hormone therapy safe and effective. At each clinic visit, measure the height and weight and calculate the growth velocity, check the IGF-1 (and adjust the dose to keep it in target), assess adherence and injection technique, examine the injection sites for lipohypertrophy, and screen for adverse effects — headache and papilloedema (benign intracranial hypertension), slipped capital femoral epiphysis, scoliosis progression, and glucose intolerance. Annual monitoring of thyroid function is wise because growth hormone can unmask central hypothyroidism. The family needs a written plan, a clear explanation of the expected response, and support for adherence, which is the single biggest determinant of outcome. [1] [2]
The definitive management of growth hormone excess begins with endoscopic transsphenoidal resection of the somatotropinoma, which is first-line and curative for many microadenomas and well-circumscribed macroadenomas. When surgery cannot achieve biochemical control — common with large, invasive or AIP-mutant tumours — medical therapy follows. A somatostatin analogue (octreotide long-acting release or lanreotide autogel) suppresses growth hormone secretion and shrinks some tumours. The growth hormone receptor antagonist pegvisomant blocks growth hormone action at the receptor and normalises IGF-1 in the majority of patients, and it has a defined role in the AIP-mutant resistant paediatric somatotropinomas that respond poorly to somatostatin analogues. Radiotherapy is reserved for residual or recurrent disease that is not controlled by surgery and medical therapy, with attention to its effects on the growth plates and on future pituitary function. [10] [11] [12]
D.O.S.E. \u2014 the rhGH therapy check
Specific Subtypes & Scenarios
Multiple pituitary hormone deficiency is the deficiency scenario that most changes management, because the child needs more than growth hormone. A PROP1, POU1F1, or HESX1 mutation, or an acquired tumour such as a craniopharyngioma, can produce combined growth hormone, thyroid, adrenal, and gonadotrophin deficiency, and the candidate must replace each axis in the correct order — hydrocortisone before thyroid hormone, and sex steroids at the appropriate pubertal age. The child with a craniopharyngioma exemplifies the trade-off: surgical resection and radiotherapy control the tumour but often leave panhypopituitarism and hypothalamic damage causing obesity, making the long-term management as much about the pituitary failure as about the tumour. [3] [10]
Turner syndrome is a licensed indication for growth hormone in the girl with short stature and a 45,X or variant karyotype, where growth hormone at the higher dose of 0.050 to 0.067 mg/kg/day improves final adult height. The Aversa systematic review confirms a height gain of several centimetres when therapy is started early and continued to near-final height, and growth hormone is now part of the standard of care alongside oestrogen replacement at puberty and surveillance for cardiac and thyroid disease. The principle is that the growth hormone is used to correct a relative growth hormone resistance and a haploinsufficiency of the SHOX gene, not an absolute deficiency. [8]
Children born small for gestational age who do not show catch-up growth by the age of two to four years are licensed for growth hormone, based on the Lee consensus and the Houk argument that early diagnosis and referral are critical for optimal growth outcomes. The dose is 0.035 to 0.067 mg/kg/day, and the candidate should know that catch-up growth is greatest in the first year of therapy and that the metabolic benefits (improved body composition) are additional to the height gain. [6] [7]
Prader-Willi syndrome carries a specific and important safety caveat. Growth hormone improves linear growth, body composition and cognitive outcomes in Prader-Willi syndrome, but these children are at risk of upper-airway obstruction, sleep apnoea and sudden death — particularly at the start of therapy and in those with pre-existing respiratory compromise or severe obesity. A sleep study and ear-nose-throat assessment should precede the start of growth hormone, the family must be warned about respiratory symptoms, and therapy should be interrupted during severe respiratory illness. The Fillion retrospective study documented both the benefits and the adverse events during growth hormone treatment in this population, and it underpins the careful selection and monitoring that is now standard. [9]
AIP-mutant familial isolated pituitary adenoma is the other genetic subtype worth naming, because somatotropinomas (and prolactinomas) arising on AIP mutations tend to present in childhood or adolescence, are often macroadenomas at diagnosis, and are relatively resistant to somatostatin analogues — making pegvisomant and aggressive surgery important, and cascade genetic testing of the family essential. [11] [10]
Complications & Pitfalls
The complications of growth hormone deficiency are partly the disease itself and partly the consequence of late or incomplete treatment. Untreated or late-treated deficiency produces a final adult height far below the genetic potential, with psychosocial consequences in adolescence and adulthood, and the metabolic actions of growth hormone deficiency contribute to an adverse body composition and an atherogenic lipid profile. In congenital hypopituitarism the complications are dominated by the associated deficiencies and the midline malformation: adrenal crisis if ACTH deficiency is unrecognised, hypoglycaemic brain injury if growth hormone deficiency is missed in the neonate, and visual impairment from septo-optic dysplasia. The complications of growth hormone excess are those of sustained IGF-1 excess and of the tumour: glucose intolerance and diabetes, hypertension, cardiomyopathy, sleep apnoea, arthropathy from acral overgrowth, dental malocclusion, visual loss from chiasmal compression, and the consequences of panhypopituitarism from tumour or its treatment. [1] [10]
The complications of recombinant growth hormone therapy are infrequent but real and must be monitored: benign intracranial hypertension (presenting with headache and papilloedema, resolving with dose reduction or cessation), slipped capital femoral epiphysis, scoliosis progression, pancreatitis, and glucose intolerance or type 2 diabetes (growth hormone antagonises insulin). The SAGhE long-term mortality study raised a signal of increased all-cause and bone-tumour mortality in adults treated with growth hormone in childhood for isolated growth hormone deficiency, idiopathic short stature and small-for-gestational-age. The absolute risk is small and the confounding is substantial, and this is precisely the kind of nuance the exam rewards. It frames growth hormone as a therapy with real benefits that must be used at the lowest effective dose for a genuine indication. [4]
Prognosis & Disposition
The prognosis of treated growth hormone deficiency is good for growth and metabolic outcomes, with most children reaching an adult height within or close to their genetic potential when therapy is started early and adherence is sustained. The response is greatest in the first year, depends on the dose and the age at starting (younger children respond better), and is reduced in children who are older, who have received cranial irradiation, or who have accompanying deficiencies. Adult height is improved but often remains below the mid-parental target, which is why early referral and a realistic conversation with the family about expectations matter. The prognosis of growth hormone excess depends on the tumour size, the genotype, the completeness of surgical resection, and the control of IGF-1; biochemical control substantially reduces the cardiovascular and metabolic mortality of acromegaly, but uncontrolled disease carries a significant excess mortality, which is the entire rationale for aggressive, lifelong management. [1] [10]
Disposition is shared, lifelong, multidisciplinary care. A paediatric endocrinologist owns the diagnosis, the stimulation or suppression testing, the initiation and titration of therapy, and the transition planning. The general paediatrician or family doctor owns the growth monitoring, the systemic screening, the coordination of the multidisciplinary team, and the front-line recognition of decompensation. The paediatric neurosurgeon, ophthalmologist, and (for the overgrowth syndromes) clinical geneticist join for the relevant subtype. The transition from paediatric to adult endocrine care at the time of final height is a high-risk moment that must be planned: the growth-hormone-deficient adolescent is re-tested and either continues adult replacement or ceases therapy, and the growth-hormone-excess young person moves into a lifelong acromegaly surveillance pathway. Every transition needs a written handover, a named adult endocrinologist, and explicit communication of the diagnosis and the surveillance plan. [3] [10]
Special Populations
The same growth hormone disorder behaves differently across populations because access, recognition and service models are unevenly distributed. In remote and Indigenous communities, later presentation, longer retrieval times, and lower rates of growth monitoring mean that growth hormone deficiency may present late with already-impaired final height potential. The practical demands of daily subcutaneous injections (cold-chain storage, supply, supervision) are also harder to sustain. So telehealth-supported shared care with a regional centre, and a clear escalation pathway for the neonate with hypoglycaemia, are disproportionately important. In migrant, refugee and asylum-seeking families, language barriers complicate the consent for recombinant growth hormone and the genetic counselling for the familial overgrowth syndromes, and an interpreter must be used for every consultation. A careful family and consanguinity history raises the pre-test probability of the genetic subtypes. [1] [12]
In adolescents transitioning to adult endocrine care, the move is the highest-risk point in the deficiency pathway: adherence to daily injections often falls, the rationale for continued therapy may seem less compelling once final height is reached, and the metabolic benefits of adult growth hormone replacement are easy to lose. In children with complex chronic disease and disability — particularly Prader-Willi syndrome with its airway risk, and the craniopharyngioma survivor with panhypopituitarism and hypothalamic obesity — fragmentation of care across endocrinology, neurosurgery, ophthalmology and respiratory medicine is the chief threat, and a written, reconciled, shared care plan with a named coordinator is the intervention that matters most. [9] [3]
In the neonate identified with congenital hypopituitarism, the immediate priority is to exclude and treat cortisol deficiency and to prevent hypoglycaemic brain injury while the diagnostic work-up proceeds; the family needs early referral to paediatric endocrinology, clear education on the sick-day hydrocortisone plan, and a coordinated magnetic resonance imaging and genetic work-up. A normal early growth pattern does not exclude a developing axis defect, and growth should be monitored with the same vigilance as in any child with a midline malformation. [3]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: the consensus guidelines for diagnosis and treatment, the natural-history and outcome studies of recombinant growth hormone therapy, and the emerging genetic and management literature on the paediatric overgrowth syndromes. The Collett-Solberg Growth Hormone Research Society international perspective (2019) is the contemporary framework for the diagnosis, genetics and therapy of short stature, integrating growth hormone deficiency with the other indications for growth hormone. The Grimberg guidelines (2016) codified the indications, dosing, and monitoring of growth hormone and IGF-1 therapy across growth hormone deficiency, idiopathic short stature and primary IGF-1 deficiency, and they remain the most widely cited operational standard. The Murray review (2016) frames the genuine controversies — the dependence of diagnosis on the cutoff and the stimulation test, the modest response in idiopathic short stature, and the safety signal — in a way that the candidate can deploy in a viva. [1] [2] [3]
The safety evidence is dominated by the SAGhE study. The Sävendahl preliminary report of Belgium, the Netherlands and Sweden (2012) documented the long-term mortality and causes of death in patients treated with recombinant growth hormone for isolated growth hormone deficiency, idiopathic short stature and small-for-gestational-age. It raised a signal of increased all-cause and bone-tumour mortality that has shaped the conservative dosing and the careful indication-setting of contemporary practice. Even so, the consensus remains that the benefit-risk balance favours treatment for genuine indications. The Guzzetti cutoff study (2016) provided the evidence base for moving the stimulation-test cutoff from 10 to 6.7 ng/mL with modern immunoassays, improving specificity. [4] [5]
The overgrowth evidence is moving fast. The Korbonits consensus on paediatric pituitary adenomas (2024) set out the diagnosis and management of the specific tumour types including somatotropinomas, and it positioned pegvisomant and the somatostatin analogues in the medical algorithm. The Joshi report of resistant paediatric somatotropinomas due to AIP mutations established the role of pegvisomant in the aggressive, somatostatin-resistant AIP-mutant phenotype, and the Daly genetic review of X-linked acrogigantism (2024) defined the GPR101-duplication genotype and its chromatin-organisation mechanism, the TADopathy. The Aversa systematic review (2024) and the Fillion Prader-Willi study (2009) anchor the indication-specific evidence for Turner syndrome and Prader-Willi syndrome respectively. [10] [11] [12] [8] [9]
In Australia and New Zealand, recombinant growth hormone is prescribed through the Pharmaceutical Benefits Scheme and the equivalent New Zealand arrangements under tightly defined criteria: proven growth hormone deficiency, Turner syndrome, Prader-Willi syndrome, chronic renal insufficiency, and small-for-gestational-age children without catch-up growth, with annual authority renewal contingent on a documented growth response. The paediatric endocrine centres in each state and at Starship in New Zealand coordinate the stimulation testing, the magnetic resonance imaging, the therapy initiation, and the transition to adult care. Pegvisomant and the somatostatin analogues are available through the specialist services for the paediatric somatotropinoma, and aeromedical retrieval to a tertiary endocrine and neurosurgical centre is the expected pathway for the macroadenoma with visual compromise or the neonate with congenital hypopituitarism and adrenal crisis. Growth monitoring through the personal health record and the universal child health schedule is the front-line screening tool, and the rural and remote workforce is supported by telehealth-linked shared care with the regional paediatric endocrine service. [1] [10]
Exam Pearls
A fellowship candidate answering on growth hormone deficiency and excess should land six anchor points and avoid three classic traps. The anchors are the growth-chart trajectory read for velocity and mid-parental distance, and the IGF-1 and IGFBP-3 as the first-tier screen with the stimulation test at the 6.7 to 10 ng/mL cutoff to confirm deficiency. Next come the recombinant growth hormone dose of 0.045 to 0.050 mg/kg/day titrated to IGF-1 (higher for Turner and small-for-gestational-age), and the oral glucose suppression test with magnetic resonance imaging for the overgrowing child. The remaining anchors are the role of transsphenoidal surgery with somatostatin analogue or pegvisomant for excess, and the Prader-Willi sleep-apnoea caveat with the transition re-test at final height. The traps are diagnosing deficiency on a single low IGF-1 without a stimulation test, missing the neonate with congenital hypopituitarism, and labelling accelerated growth a normal variant without checking the IGF-1. The candidate who can name the modern genetic subtypes — AIP and X-linked acrogigantism — and frame the SAGhE safety signal with appropriate nuance will separate from the field. [1] [2] [10] [4]
References
- [1]Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS, et al. Diagnosis, genetics, and therapy of short stature in children: a Growth Hormone Research Society international perspective. Horm Res Paediatr, 2019.PMID 31514194
- [2]Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, et al. Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr, 2016.PMID 27884013
- [3]Murray PG, Dattani MT, Clayton PE. Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence. Arch Dis Child, 2016.PMID 26153506
- [4]Sävendahl L, Cooke D, Tidblad A, Beckers D, Butler S, Cianfarani S, et al. Long-term mortality and causes of death in isolated GHD, ISS, and SGA patients treated with recombinant growth hormone during childhood in Belgium, The Netherlands, and Sweden: preliminary report of 3 countries participating in the EU SAGhE study. J Clin Endocrinol Metab, 2012.PMID 22238393
- [5]Guzzetti C, Ibba A, Pilia S, Beltrami N, Loche S. Cut-off limits of the peak GH response to stimulation tests for the diagnosis of GH deficiency in children and adolescents: study in patients with organic GHD. Eur J Endocrinol, 2016.PMID 27147639
- [6]Lee PA, Chernausek SD, Hokken-Koelega AC, Czernichow P; International Small for Gestational Age Advisory Board. International Small for Gestational Age Advisory Board consensus development conference statement: management of short children born small for gestational age, April 24-October 1, 2001. Pediatrics, 2003.PMID 12777538
- [7]Houk CP, Lee PA. Early diagnosis and treatment referral of children born small for gestational age without catch-up growth are critical for optimal growth outcomes. Int J Pediatr Endocrinol, 2012.PMID 22559301
- [8]Aversa T, Valenzise M, De Leonibus E, Gallizzi R, Salzano G, Sferlazzas C, et al. Growth hormone treatment to final height in Turner syndrome: systematic review. Clin Ther, 2024.PMID 38151406
- [9]Fillion M, Deal C, Van Vliet G. Retrospective study of the potential benefits and adverse events during growth hormone treatment in children with Prader-Willi syndrome. J Pediatr, 2009.PMID 18814886
- [10]Korbonits M, Akker SA, Leontiou CA, Ferraù F, Pivonello R, Jacobson DA, et al. Consensus guideline for the diagnosis and management of pituitary adenomas in childhood and adolescence: Part 2, specific diseases. Nat Rev Endocrinol, 2024.PMID 38336898
- [11]Joshi K, Cory M, Yuen KC, Dal J, Trifiro G, Hwa V, et al. Resistant paediatric somatotropinomas due to AIP mutations: role of pegvisomant. Horm Res Paediatr, 2018.PMID 29953972
- [12]Daly AF, Yuan B, Ferraù F, Auriemma RS, Biarmes-Gomez X, Beckers A. The genetic pathophysiology and clinical management of the TADopathy, X-linked acrogigantism. Endocr Rev, 2024.PMID 38696651