Paeds · endocrinology-diabetes-and-growth
Hypercalcaemia and hyperparathyroidism
Also known as Hypercalcaemia · Hypercalcemia · Primary hyperparathyroidism · Paediatric hyperparathyroidism · Familial hypocalciuric hypercalcaemia · Neonatal severe hyperparathyroidism
Fellowship guide to paediatric hypercalcaemia and hyperparathyroidism: the PTH fork that sorts every high calcium, the single test (urine calcium-to-creatinine clearance ratio) that stops you operating on familial hypocalciuric hypercalcaemia, the saline-first acute resuscitation, and the calcitonin-then-bisphosphonate sequence that locks calcium into bone.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single test that organises everything in paediatric hypercalcaemia is the parathyroid hormone level. When calcium is high, a normal parathyroid axis switches PTH off. So a PTH that has not switched off is the signature of a parathyroid-driven process, and a PTH that has switched off sends you looking elsewhere. Holding that fork in mind turns a long differential into two short lists and a clear plan. [1] [6]
This page covers the recognition and management of hypercalcaemia and hyperparathyroidism in children: the PTH-based classification that splits every high calcium, the urine calcium-to-creatinine clearance ratio that separates familial hypocalciuric hypercalcaemia from primary hyperparathyroidism, the saline-first resuscitation of hypercalcaemic crisis, the calcitonin-then-bisphosphonate drug sequence, the hereditary syndromes (MEN1, hyperparathyroid-jaw tumour, neonatal severe hyperparathyroidism), and the surgical pathway for parathyroidectomy. It links to the calcium-magnesium-phosphate leaf for broader electrolyte detail. [1] [7]
Overview & Definition
Hypercalcaemia is a serum calcium above the age-appropriate reference range, taken as an adjusted (albumin-corrected) calcium above 2.6 mmol/L or an ionised calcium above 1.3 mmol/L. The total calcium must be corrected for albumin, because nearly half of circulating calcium is protein-bound and a low albumin hides a high ionised fraction: add 0.02 mmol per litre for every gram per litre that the albumin sits below forty. [6]
What makes paediatric hypercalcaemia distinctive is that it is uncommon and that the causes cluster by age. Neonates present with inherited calcium-sensing receptor disorders, Williams syndrome, subcutaneous fat necrosis and iatrogenic causes. School-age children and adolescents more often have primary hyperparathyroidism or vitamin D intoxication. The threshold for action is the same regardless of cause: a symptomatic child or a calcium above 3.5 mmol per litre is an emergency, because the rising calcium poisons renal concentrating ability, depresses the central nervous system and shortens the cardiac QT interval. [6] [10]
Classification
Classify hypercalcaemia by the parathyroid hormone response, because the PTH level sends you down one of two distinct investigative and therapeutic paths. The figure below splits a high calcium into PTH-dependent disease, where the parathyroid gland is the driver, and PTH-independent disease, where the calcium comes from bone, gut, kidney or a tumour and the suppressed parathyroids are bystanders. [1]
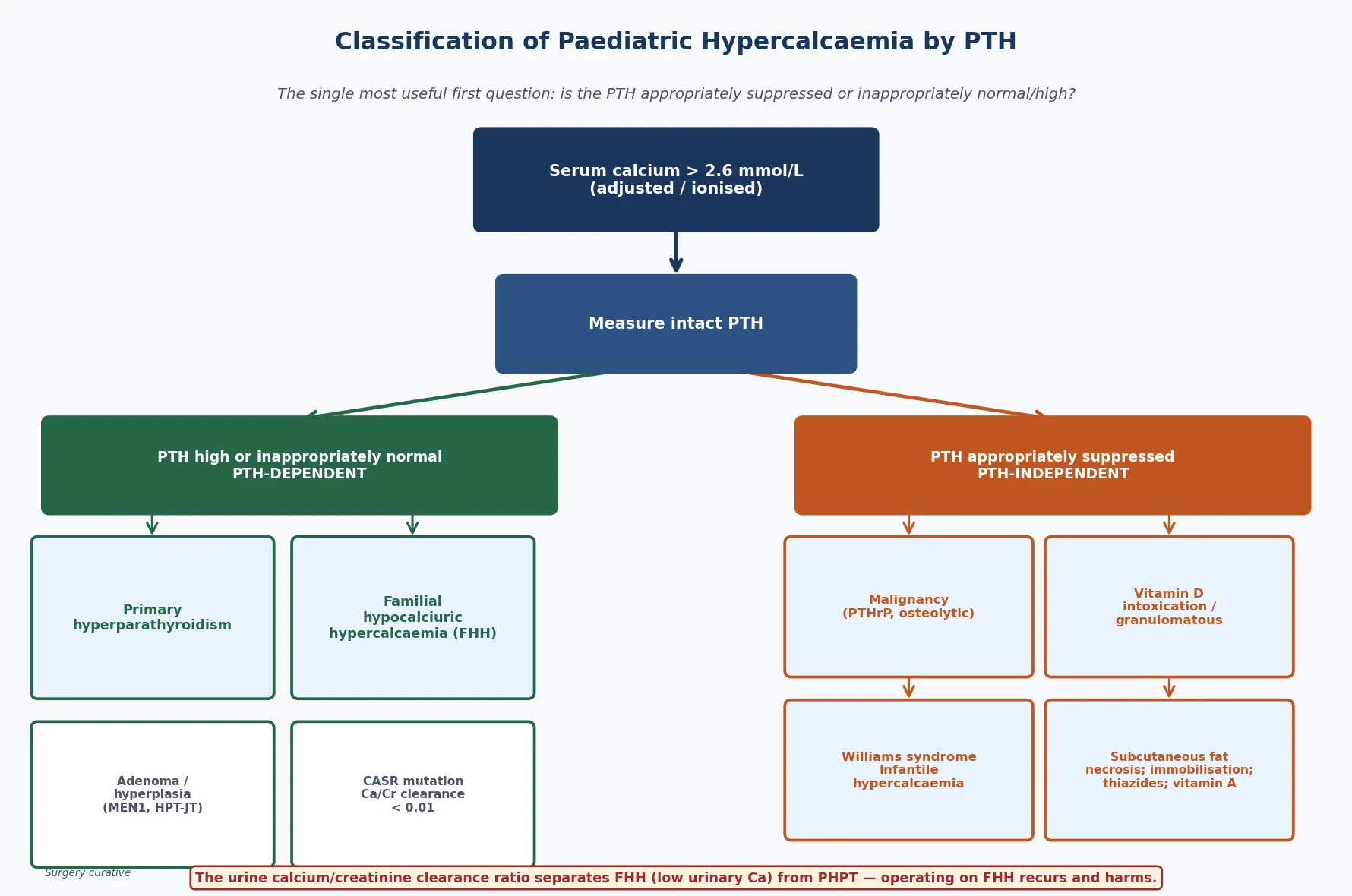
PTH-dependent (high or inappropriately normal PTH)
- Primary hyperparathyroidism — single adenoma or multigland hyperplasia
- Familial hypocalciuric hypercalcaemia (FHH) — CASR mutation, benign
- Neonatal severe hyperparathyroidism (NSHPT) — homozygous CASR
- Hereditary syndromes: MEN1, MEN2A, MEN4, hyperparathyroid-jaw tumour
PTH-independent (suppressed PTH)
- Malignancy — PTHrP secretion, osteolytic metastases, tumour lysis
- Vitamin D intoxication and granulomatous disease (excess 1,25-dihydroxyvitamin D)
- Williams syndrome and idiopathic infantile hypercalcaemia
- Subcutaneous fat necrosis, immobilisation, thiazides, vitamin A, milk-alkali
The age of the child reshuffles the probabilities within each arm. A neonate with a high calcium and an inappropriately normal PTH is most likely to have familial hypocalciuric hypercalcaemia or its severe homozygous form, neonatal severe hyperparathyroidism. A school-age child or adolescent in the same arm is more likely to have a parathyroid adenoma, sometimes as part of a multiple endocrine neoplasia syndrome. In the PTH-independent arm, infants point toward Williams syndrome, subcutaneous fat necrosis and excess vitamin D, while older children point toward malignancy and immobilisation. [5] [6]
Epidemiology & Risk Factors
Primary hyperparathyroidism is far rarer in children than in adults, where it is a disease of postmenopausal women. The paediatric incidence is roughly two to five per hundred thousand per year, and most paediatric series report a slight female predominance and a mean age at presentation in the early teenage years. Children are also more likely than adults to be symptomatic at presentation, with stones, bone pain or nonspecific abdominal and neuropsychiatric complaints, because the diagnosis is rarely caught by an incidental screening blood test. [5]
The strongest risk factor for a parathyroid-driven cause is a hereditary endocrine tumour predisposition syndrome. Multiple endocrine neoplasia type 1 carries parathyroid hyperplasia together with pancreatic and pituitary tumours, and parathyroid disease is its commonest and earliest expression. The hyperparathyroid-jaw tumour syndrome (CDC73, formerly HRPT2) pairs parathyroid disease with ossifying jaw fibromas and carries a risk of parathyroid carcinoma. A family history of early-onset hypercalcaemia, kidney stones or endocrine tumours lowers the threshold to test and to screen relatives. [7] [8]
CALCIUM
Fatigue, irritability, depression, and at high levels seizures and coma — calcium depresses the central nervous system
Constipation, nausea, anorexia, and pancreatitis from smooth-muscle hypotonia and calcium deposition
Nephrolithiasis and nephrocalcinosis from chronic hypercalciuria; polyuria from nephrogenic diabetes insipidus
Shortened QT interval on the ECG, and at very high levels arrhythmia
A suppressed PTH points to PTH-independent causes — malignancy, vitamin D, drugs, bone
The calcium-to-creatinine clearance ratio separates FHH (below 0.01) from primary hyperparathyroidism
Saline first, then calcitonin and bisphosphonate, then treat the cause; never operate on FHH
The reason vitamin D excess tops the PTH-independent list in some paediatric populations is the widespread and increasingly unregulated use of high-dose vitamin D supplements, both prescribed and over-the-counter. A careful supplement and medication history — including parent-administered preparations, herbal remedies and fortification practices — is the first investigation of any PTH-independent hypercalcaemia, because identifying and stopping the source is the cure. [6]
Pathophysiology
To see why calcium runs out of control, picture the calcium homeostasis loop as a thermostat. The calcium-sensing receptor on the surface of parathyroid cells monitors the blood calcium and switches off parathyroid hormone secretion when calcium is high. The parathyroid hormone then acts on bone, kidney and gut to raise calcium, completing the feedback loop. When the thermostat works, a high calcium switches the hormone off and the calcium settles. [7] [9]

Two mechanisms matter at the bedside. First, when an autonomous parathyroid adenoma secretes hormone regardless of the calcium level, the high hormone drives bone resorption, renal calcium reabsorption and vitamin D activation, so the calcium climbs while the phosphate falls — the classic biochemical pair of primary hyperparathyroidism. Second, when the calcium-sensing receptor is defective, as in familial hypocalciuric hypercalcaemia, the parathyroid cell under-reads the calcium and keeps the hormone mildly raised, while the kidney tubule over-reabsorbs calcium so the urine calcium is low. [8] [9]
The PTH-independent causes work downstream of the parathyroid gland, which correctly switches off. Malignancy raises calcium through parathyroid-hormone-related protein, a tumour product that mimics the bone and kidney effects of parathyroid hormone without being detected by the PTH assay. Vitamin D intoxication and granulomatous disease generate excess 1,25-dihydroxyvitamin D, which drives gut calcium absorption. Immobilisation releases calcium from unopposed bone resorption, and subcutaneous fat necrosis releases prostaglandins and calcium from necrotic fat. Recognising which pathway is at work guides both the acute therapy and the definitive plan. [6] [10]
Clinical Presentation
A child with mild hypercalcaemia is often asymptomatic and the finding is incidental, discovered on a blood test taken for another reason. As the calcium climbs, the child becomes thirsty and passes a lot of urine, because the high calcium impairs renal concentrating ability and produces a partial nephrogenic diabetes insipidus. Polyuria and polydipsia are early and useful clues, and they explain why a hypercalcaemic crisis presents with dehydration. [5] [6]
The constellation of symptoms is captured by the stones, bones, abdominal groans and psychic moans mnemonic. Kidney stones and nephrocalcinosis reflect chronic hypercalciuria. Bone pain, and rarely fractures or brown tumours, reflect parathyroid-hormone-driven resorption in hyperparathyroidism. The abdominal groans are anorexia, nausea, constipation and occasionally pancreatitis from depressed smooth-muscle tone and calcium deposition. The psychic moans are fatigue, irritability, low mood and impaired concentration, which are easy to dismiss in a teenager until the calcium is found. [5]
The neonate with severe hyperparathyroidism presents a different and dramatic picture. The infant is hypotonic, feeds poorly, breathes rapidly and may have fractures, and the calcium is markedly elevated. This is usually neonatal severe hyperparathyroidism from homozygous or compound-heterozygous calcium-sensing receptor mutations, and it is life-threatening without prompt recognition and parathyroidectomy. Any neonate with an unexplained high calcium and respiratory distress has this until proven otherwise. [6] [9]
Differential Diagnosis
The differential turns on the PTH level, which is why it is the first test after confirming the calcium. A high or inappropriately normal PTH with hypercalcaemia narrows the field to parathyroid-driven disease, and within that group the urine calcium-to-creatinine clearance ratio separates the two conditions that look alike but are managed oppositely. [1] [8]
Primary hyperparathyroidism
- Single parathyroid adenoma in roughly 80% of paediatric cases
- Multigland hyperplasia in MEN1, MEN2A, MEN4 — screen the family
- Hyperparathyroid-jaw tumour syndrome (CDC73) — risk of carcinoma
- Urine calcium-to-creatinine clearance ratio above 0.01; high urinary calcium
Familial hypocalciuric hypercalcaemia
- Calcium-sensing receptor loss-of-function mutation (CASR)
- Mild lifelong hypercalcaemia, asymptomatic, low urinary calcium
- Calcium-to-creatinine clearance ratio below 0.01
- Benign — parathyroidectomy recurs and causes hypocalcaemia
The ratio is calculated from paired serum and urine calcium and creatinine values, and a result below 0.01 points to familial hypocalciuric hypercalcaemia. This matters because the two conditions are managed in opposite directions: primary hyperparathyroidism is cured by parathyroidectomy, while operating on familial hypocalciuric hypercalcaemia removes normal tissue, the calcium recurs, and the child is left hypocalcaemic and on lifelong calcium and vitamin D replacement. Confirming the ratio, and ideally a CASR gene test, before any surgery is the habit that prevents the wrong operation. [9]
For PTH-independent causes, the history and a few targeted tests sort the list. Malignancy is suggested by weight loss, anaemia and a high parathyroid-hormone-related protein or evidence of an osteolytic lesion. Vitamin D intoxication shows a high 25-hydroxyvitamin D and a high 1,25-dihydroxyvitamin D, while granulomatous disease shows a normal 25-hydroxyvitamin D but a high 1,25-dihydroxyvitamin D. Williams syndrome carries its elfin facies, supravalvular aortic stenosis and developmental features, and subcutaneous fat necrosis shows its firm back and buttock nodules in a recent neonate. [6] [10]
Clinical & Bedside Assessment
The focused assessment of a hypercalcaemic child rests on three observations that take minutes but settle the urgency: assess the hydration and consciousness, examine the abdomen and skin, and look for the syndromic features that point to a hereditary cause. A child who is dry, vomiting, confused or hypotonic is in crisis and needs immediate intravenous access and saline. [6]
Examine the abdomen for constipation and the rare pancreatitis, and palpate carefully for an abdominal mass that might signal a malignancy. Inspect the skin and subcutaneous tissue of a neonate for the firm indurated plaques of subcutaneous fat necrosis, which typically appear over the back, shoulders and buttocks in the weeks after a hypoxic or traumatic delivery. Look for the dysmorphic features of Williams syndrome — the elfin facies, periorbital fullness and stellate iris pattern — and auscultate for the supravalvular aortic stenosis murmur that accompanies it. [6]
The assessment is rounded out by asking about supplements and medications. Specifically ask about vitamin D preparations, including high-dose stoss therapy, over-the-counter and herbal preparations, vitamin A or retinoid therapy, thiazide diuretics, calcium-based antacids and lithium. A family history of hypercalcaemia, kidney stones, parathyroid surgery or endocrine tumours raises the hereditary syndromes and is worth taking in the parents as well as the child. These features, taken with the corrected calcium, justify the targeted biochemical panel rather than repeated calcium measurements. [5] [8]
Investigations
The investigation pathway proceeds in two stages: confirm the calcium, then find its source through the PTH fork. Always confirm a high total calcium with an albumin correction or, ideally, an ionised calcium, because a high total calcium with a low albumin may be artefactual. A repeat measurement, drawn fasting and without a tourniquet, settles spurious elevations from haemoconcentration. [1]
Once the calcium is confirmed, measure intact parathyroid hormone drawn at the same time as the calcium. A high or inappropriately normal hormone level means PTH-dependent disease; a suppressed level means PTH-independent disease. This single result splits the investigation into two paths. For PTH-dependent disease, measure a spot urine calcium and creatinine alongside serum values to calculate the calcium-to-creatinine clearance ratio, which separates primary hyperparathyroidism from familial hypocalciuric hypercalcaemia. [3] [9]
PTH-dependent work-up
- Intact PTH high or inappropriately normal
- Calcium-to-creatinine clearance ratio: above 0.01 = PHPT, below 0.01 = FHH
- Serum phosphate low, vitamin D 25-OH checked and replaced before surgery
- Parathyroid imaging (sestamibi SPECT, ultrasound) and CASR / MEN1 genetics
PTH-independent work-up
- Intact PTH suppressed — confirm the calcium is genuinely high
- PTHrP for malignancy; 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D
- Renal function, phosphate, alkaline phosphatase, thyroid function
- Imaging for malignancy; review drugs (vitamin A, thiazides, lithium, calcium)
For confirmed primary hyperparathyroidism, localise the gland before surgery with a technetium-99m sestamibi single-photon emission computed tomography scan and a high-resolution neck ultrasound, which together identify a single adenoma in most children. Genetic testing for the multiple endocrine neoplasia genes and the calcium-sensing receptor guides the surgical extent and the family screening, because multigland hyperplasia needs a different operation (subtotal or total parathyroidectomy) from a single adenoma (focused removal). Always correct any vitamin D deficiency before parathyroid surgery to limit postoperative hungry bone syndrome. [5] [8]
Management — Resuscitation
Severe or symptomatic hypercalcaemia is a medical emergency and the first intervention is always volume. The rising calcium impairs renal concentrating ability and produces polyuria, while the gastrointestinal effects produce vomiting, so the child arrives volume-depleted and the dehydration itself worsens the calcium through reduced glomerular filtration and calcium retention. Replacing the volume is both resuscitation and treatment. [4] [6]
Give aggressive isotonic saline, at ten to twenty millilitres per kilogram as an initial bolus over about one hour if volume depleted, then maintenance in the first hours and titrated to euvolaemia, with careful monitoring of electrolytes and fluid balance. Once the child is euvolaemic, add calcitonin at four units per kilogram subcutaneously or intravenously every twelve hours, which lowers calcium within hours by inhibiting osteoclast activity; its effect is rapid but tachyphylactic, lasting only a few days. Follow with a bisphosphonate such as pamidronate or zoledronic acid, which takes two to four days to work but provides a durable fall in calcium. [4] [10]
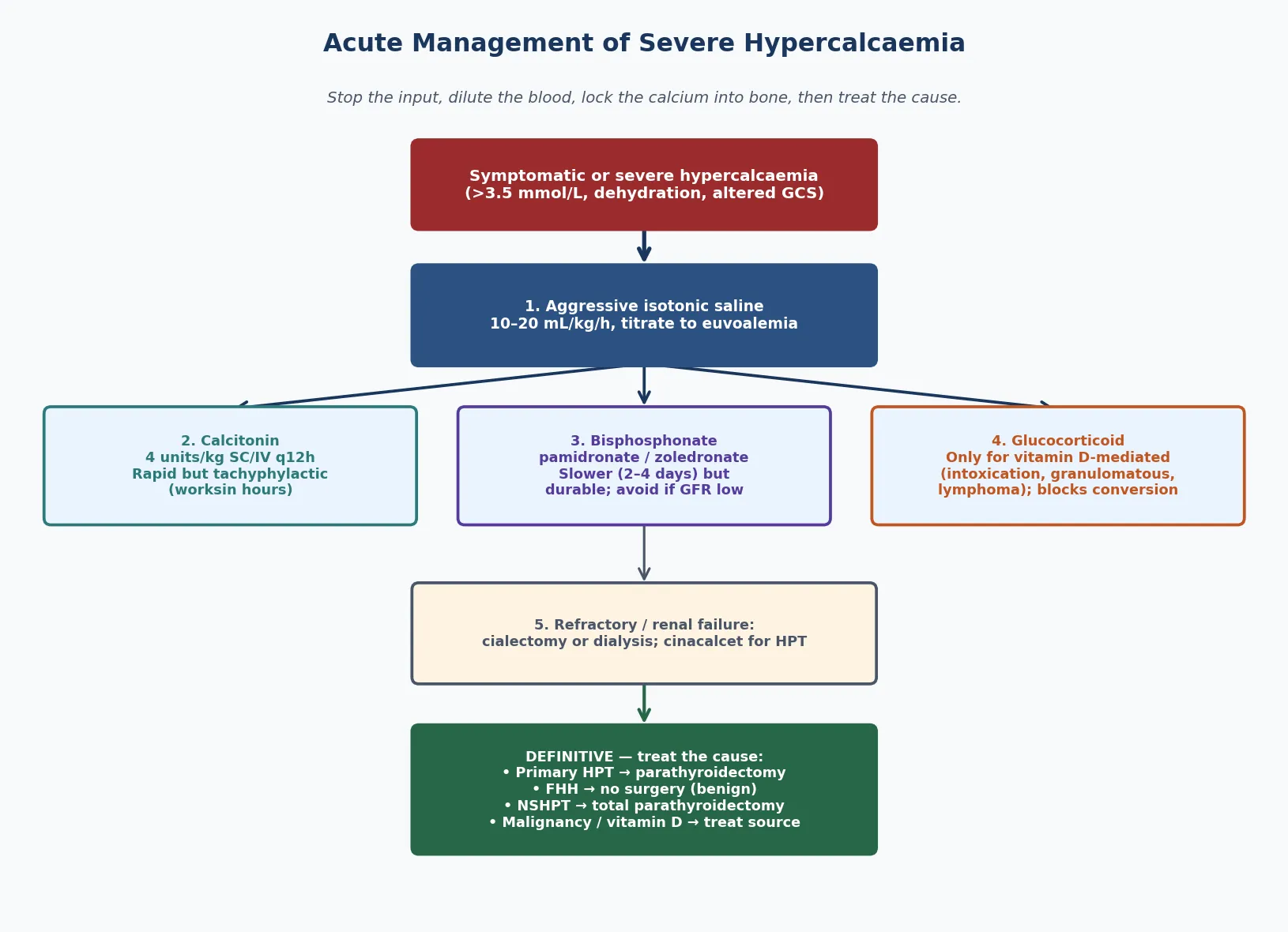
For the small group refractory to saline, calcitonin and bisphosphonate, or for the child in renal failure where bisphosphonates are nephrotoxic, the options are haemodialysis or peritoneal dialysis to remove calcium directly, and cinacalcet, a calcimimetic that sensitises the calcium-sensing receptor and is useful in parathyroid-driven disease. Glucocorticoids have a specific and limited role: they lower calcium only when vitamin D is the driver, because they block the one-alpha-hydroxylation of vitamin D in the kidney and macrophages. They work for vitamin D intoxication and granulomatous disease, and they do not work for primary hyperparathyroidism or parathyroid-hormone-related protein-mediated malignancy. [4] [10]
Management — Definitive & Stepwise
Definitive treatment is dictated by the cause. For primary hyperparathyroidism, the treatment is surgical removal of the abnormal gland or glands, and in children — who are more often symptomatic — surgery is offered more liberally than in adults. A single adenoma is removed by a focused parathyroidectomy guided by preoperative localisation and intraoperative parathyroid hormone monitoring, in which a hormone fall of more than fifty percent into the normal range at ten minutes confirms cure. Multigland hyperplasia, as in multiple endocrine neoplasia, needs a subtotal or total parathyroidectomy with autotransplantation. [5] [8]
For familial hypocalciuric hypercalcaemia, the definitive management is no surgery. The condition is benign and lifelong, the calcium is stable and usually asymptomatic, and parathyroidectomy does not cure it — the calcium recurs and the child is left hypocalcaemic. The task is to make the diagnosis, document the calcium-sensing receptor mutation, reassure the family, and protect the child from unnecessary parathyroid exploration. Screen relatives, because the condition is autosomal dominant and identifying affected family members prevents the same mistake in them. [9]
For neonatal severe hyperparathyroidism, the homozygous calcium-sensing receptor disorder, medical therapy with a calcimimetic and bisphosphonate may stabilise the calcium, but the disease is often life-threatening and a total parathyroidectomy is usually required. For PTH-independent causes, the definitive treatment is to remove the driver: stop the vitamin D supplement, treat the malignancy, mobilise the immobilised child, excise the granulomatous tissue, or stop the offending drug. Denosumab, a receptor activator of nuclear factor kappa-B ligand inhibitor, has an emerging role in refractory malignancy-associated and immobilisation hypercalcaemia in children where bisphosphonates have failed or are unsuitable. [6] [10]
Monitoring is the long game. Track the serum calcium and phosphate, the renal function and the bone mineral density, and after parathyroid surgery watch closely for hypocalcaemia from hungry bone syndrome, in which the sudden removal of parathyroid hormone drives calcium into the hungry, demineralised bone. Give oral calcium and calcitriol, and intravenous calcium if severe, and taper as the skeleton remineralises over weeks. Lifelong surveillance for the hereditary syndromes, and a planned transition to adult endocrinology, complete the pathway. [5] [8]
Specific Subtypes & Scenarios
Primary hyperparathyroidism in an adolescent is the archetype of the PTH-dependent arm. A single parathyroid adenoma drives a high calcium with a high or inappropriately normal hormone, a low phosphate and a high urinary calcium, and the child presents with stones, bone pain or nonspecific symptoms. Focused parathyroidectomy with intraoperative hormone monitoring is curative, and screening for multiple endocrine neoplasia is part of the work-up because the syndromic form needs a different operation. [5]
Familial hypocalciuric hypercalcaemia is the great mimic and the great trap. It presents with a mild, stable, lifelong hypercalcaemia, an inappropriately normal parathyroid hormone and, crucially, a low urinary calcium with a calcium-to-creatinine clearance ratio below 0.01. The child is well, the family has a history of "high calcium that runs in the family," and the danger is the unnecessary operation. A calcium-sensing receptor gene test confirms the diagnosis, and the management is recognition and reassurance, not surgery. [9]
Neonatal severe hyperparathyroidism is the homozygous or compound-heterozygous form of the same receptor defect, and it is the most dramatic presentation of hypercalcaemia in children. The neonate is profoundly hypercalcaemic, hypotonic, breathless and may have fractures, and the disease is fatal without treatment. Medical therapy with a calcimimetic, calcitonin and bisphosphonate may buy time, but a total parathyroidectomy with lifelong calcium and calcitriol replacement is usually needed. Hypercalcaemia of malignancy, driven by parathyroid-hormone-related protein or osteolysis, is rare in children but enters the differential of a PTH-independent high calcium with weight loss and bony pain. [6] [9]
Williams syndrome and vitamin D intoxication round out the paediatric-specific scenarios. Williams syndrome produces infantile hypercalcaemia alongside its elfin facies, supravalvular aortic stenosis, friendly personality and developmental delay, and the hypercalcaemia usually resolves with a low-calcium and low-vitamin-D diet. Vitamin D intoxication, whether from prescribed high-dose therapy, over-the-counter supplements or fortification error, produces a high 25-hydroxyvitamin D, and the management is to stop the source, give saline and glucocorticoids, and support the child until the fat-soluble vitamin clears. [6]
Complications & Pitfalls
The untreated disease carries a heavy burden. Chronic hypercalcaemia causes nephrocalcinosis and kidney stones, and in hyperparathyroidism the bone resorption produces osteopenia, fractures and brown tumours. The neuropsychiatric toll — fatigue, low mood, impaired concentration — is easy to underestimate, and the shortened QT interval can precipitate arrhythmia at very high levels. Many of these are partly reversible with cure, but renal and skeletal damage may persist. [5] [6]
The treatment carries its own complications. Hungry bone syndrome after parathyroidectomy drives calcium into the demineralised skeleton and produces severe, sometimes symptomatic hypocalcaemia within hours to days, needing intravenous calcium and calcitriol. Bisphosphonate nephrotoxicity limits the use of these drugs in renal failure, and calcitonin tachyphylaxis means its rapid effect is short-lived. Over-treatment with saline risks fluid overload, and glucocorticoids used for the wrong cause (primary hyperparathyroidism or parathyroid-hormone-related protein-mediated malignancy) simply do not work and carry their own burden. [4] [10]
The classic pitfall is the mislabelled familial hypocalciuric hypercalcaemia. Operating on this benign condition recurs and harms, and the error is preventable by calculating the urine calcium-to-creatinine clearance ratio before any neck exploration. The converse error — missing a symptomatic primary hyperparathyroidism because the parathyroid hormone was not measured, or attributing the high calcium to a supplement the child does not take — delays the curative surgery. Reaching for the parathyroid hormone and the urine calcium ratio before the operating list is the habit that prevents both. [8] [9]
Prognosis & Disposition
With expert parathyroidectomy, primary hyperparathyroidism is cured in the great majority of children, and catch-up bone mineralisation usually follows. Final bone density and renal outcome depend on the duration and severity of the disease before treatment, which is why earlier diagnosis is the single biggest modifiable prognostic factor. Recurrence is rare after a successful single-gland removal, but the hereditary syndromes carry a lifelong risk of multigland recurrence that demands structured surveillance. [5] [8]
Familial hypocalciuric hypercalcaemia carries an excellent prognosis because it is benign, stable and asymptomatic, provided the child is protected from unnecessary surgery. Neonatal severe hyperparathyroidism is life-threatening and its prognosis depends on the speed of recognition and definitive surgery. The PTH-independent causes inherit the prognosis of their driver: malignancy-associated hypercalcaemia reflects advanced tumour burden, while vitamin D intoxication and Williams syndrome resolve with removal of the source or time. Disposition is to a specialist paediatric endocrine service with experienced parathyroid surgical expertise, and a planned transition to adult endocrinology for the hereditary conditions. [6] [7]
Special Populations
Neonates are the population in which hypercalcaemia is most dangerous and most often inherited. Neonatal severe hyperparathyroidism, Williams syndrome, subcutaneous fat necrosis and iatrogenic causes dominate, and the work-up leans toward calcium-sensing receptor genetics, vitamin D metabolites and a careful perinatal and supplement history. A neonate with severe hypercalcaemia and respiratory distress needs urgent endocrine and surgical input, not watchful waiting. [6] [9]
Adolescents with primary hyperparathyroidism face the added burden of the hereditary syndromes, body-image distress from bone disease and the disruption of stones and abdominal pain. They need honest counselling about the operation, the postoperative calcium replacement, and the implications of a multiple endocrine neoplasia diagnosis for their own future and their family. Children with complex chronic illness and immobilisation are a growing population at risk of immobilisation hypercalcaemia, in which unopposed bone resorption drives the calcium up, and the management is mobilisation where possible alongside the standard drug sequence. [5] [10]
Indigenous, migrant and remote-dwelling families may face the logistics of accessing a specialist parathyroid-surgical service, and the supplement and dietary practices that drive vitamin D intoxication vary with cultural background. A clear local pathway, telehealth support, a written emergency plan that travels with the child, and access to interpreter services are essential so that the diagnosis and the definitive operation are not delayed by geography or language. [3]
Evidence, Guidelines & Regional Differences
The evidence base is anchored by the Fifth International Workshop guidelines on primary hyperparathyroidism, which updated the Fourth Workshop summary and expanded the role of surgery and skeletal and renal assessment. The Canadian and international consensus of Khan and colleagues complements these guidelines with practical recommendations on evaluation and management, and the medical management proceedings of Marcocci and colleagues from the Fourth Workshop set out the drug options. The paediatric-specific evidence draws on the Mayo Clinic paediatric series of Kollars and colleagues, which remains the defining cohort. [1] [3] [4]
The genetic and neonatal evidence draws on the historical review of Marx and Goltzman on the hyperparathyroid syndromes, the hereditary syndromes review of Marx and colleagues, the genetic causes of neonatal and infantile hypercalcaemia by Gorvin, and the calcium-sensing receptor mutation work of Pidasheva and colleagues. The emerging therapy evidence for refractory paediatric hypercalcaemia, including the multicentre denosumab experience of Hobbs and colleagues, rounds out the contemporary picture. [6] [7] [10]
Regional practice differences are modest because the International Workshop guidelines are adopted across Australia, New Zealand and internationally, but the threshold for surgery, the availability of paediatric parathyroid surgeons and intraoperative parathyroid hormone monitoring, and access to calcimimetics and newer agents vary between centres. In Australia and New Zealand, suspected paediatric hyperparathyroidism is referred to a tertiary paediatric endocrine service with a linked surgical and nuclear-medicine team, reflecting the centralised expertise that the evidence supports. The main controversies are the role of cinacalcet in children awaiting surgery, the optimal management of mild asymptomatic hyperparathyroidism in adolescence, and the long-term safety of bisphosphonates and denosumab in the growing skeleton. [1] [4]
Exam Pearls
Hold one sentence for the viva: when you find a high calcium, measure the parathyroid hormone first, because a high or inappropriately normal hormone means parathyroid-driven disease and a suppressed hormone sends you looking elsewhere. The single test that prevents the wrong operation is the urine calcium-to-creatinine clearance ratio, which separates familial hypocalciuric hypercalcaemia below 0.01 from primary hyperparathyroidism above it. [1] [9]
State the frequently misremembered facts correctly. Glucocorticoids only work when vitamin D is the driver — intoxication, granulomatous disease, some lymphomas — and they do not work for primary hyperparathyroidism or parathyroid-hormone-related protein-mediated malignancy. The crisis threshold is a corrected calcium above 3.5 mmol per litre, and the first intervention is aggressive isotonic saline at ten to twenty millilitres per kilogram as an initial bolus over about one hour if volume depleted, then maintenance, because you cannot lower calcium in a dehydrated child. The calcium-to-creatinine clearance ratio cut-off is 0.01. And calcitonin works fast but tachyphylactic, while bisphosphonates work slowly but durably. [4] [6]
The high-yield lesion-sign pairings: an adolescent with stones, bone pain and a high parathyroid hormone has primary hyperparathyroidism (a single adenoma); a well child with a mild stable calcium, a family history of high calcium and a low urine calcium has familial hypocalciuric hypercalcaemia (do not operate); a hypotonic neonate with a markedly high calcium and respiratory distress has neonatal severe hyperparathyroidism; and a child with elfin facies, a murmur and hypercalcaemia has Williams syndrome. These pairings do most of the diagnostic work in the short case. [5] [6]
References
- [1]Bilezikian JP; Khan AA; Silverberg SJ; Fuleihan GE; et al Evaluation and Management of Primary Hyperparathyroidism: Summary Statement and Guidelines from the Fifth International Workshop. J Bone Miner Res, 2022.PMID 36245251
- [2]Bilezikian JP; Brandi ML; Eastell R; Silverberg SJ; et al Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab, 2014.PMID 25162665
- [3]Khan AA; Hanley DA; Rizzoli R; Bollerslev J; et al Primary hyperparathyroidism: review and recommendations on evaluation, diagnosis, and management. A Canadian and international consensus. Osteoporos Int, 2017.PMID 27613721
- [4]Marcocci C; Bollerslev J; Khan AA; Shoback DM Medical management of primary hyperparathyroidism: proceedings of the fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism. J Clin Endocrinol Metab, 2014.PMID 25162668
- [5]Kollars J; Zarroug AE; van Heerden J; Lteif A; et al Primary hyperparathyroidism in pediatric patients. Pediatrics, 2005.PMID 15805373
- [6]Gorvin CM Genetic causes of neonatal and infantile hypercalcaemia. Pediatr Nephrol, 2022.PMID 33990852
- [7]Marx SJ; Goltzman D Evolution of Our Understanding of the Hyperparathyroid Syndromes: A Historical Perspective. J Bone Miner Res, 2019.PMID 30536424
- [8]Marx SJ; Simonds WF; Agarwal SK; Burns AL; et al Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Miner Res, 2002.PMID 12412776
- [9]Pidasheva S; Canaff L; Simonds WF; Marx SJ; et al Impaired cotranslational processing of the calcium-sensing receptor due to signal peptide missense mutations in familial hypocalciuric hypercalcemia. Hum Mol Genet, 2005.PMID 15879434
- [10]Hobbs A; Nair R; Ludwig K; Ambler GR; et al Denosumab as a treatment for pediatric hypercalcemia-a multicenter experience. JBMR Plus, 2026.PMID 41522662