Paeds · endocrinology-diabetes-and-growth
Hypocalcaemia and hypoparathyroidism
Also known as Hypocalcaemia · Hypoparathyroidism · Pseudohypoparathyroidism · DiGeorge syndrome hypocalcaemia · Neonatal hypocalcaemia · Calcium homeostasis disorder · PTH resistance · 22q11.2 deletion hypocalcaemia
Fellowship guide to hypocalcaemia and hypoparathyroidism in children: the calcium-PTH axis from parathyroid chief cell to bone, kidney and gut, neonatal versus later-onset hypocalcaemia, the PTH-led classification that splits every cause, acute symptomatic tetany and seizure management with IV calcium gluconate, and the long-term trio of oral calcium, calcitriol and PTH 1-34 for refractory disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The organising principle is the calcium–PTH axis: the parathyroid calcium-sensing receptor detects a fall in ionised calcium, the chief cells release parathyroid hormone, and PTH restores calcium by mobilising bone, reabsorbing calcium in the kidney, and — indirectly through activated vitamin D — increasing gut absorption. Hypoparathyroidism breaks this axis at the source: no PTH is made. Pseudohypoparathyroidism breaks it at the target organ: PTH is made in abundance but the kidney and bone cannot respond. Vitamin D deficiency breaks it at the gut. Knowing where the axis breaks is the fastest route to the right diagnosis and treatment. [3] [8]
This page covers the full breadth of paediatric hypocalcaemia: the physiology of calcium homeostasis, the PTH-led classification, neonatal early- and late-onset hypocalcaemia, 22q11.2 deletion (DiGeorge) syndrome, autoimmune polyglandular syndrome type 1, pseudohypoparathyroidism, acute symptomatic management with intravenous calcium, and the long-term trio of oral calcium, calcitriol and PTH 1-34. It cross-links to the rickets and metabolic bone disease leaf rather than repeating the full vitamin D pathway. [1] [7]
Overview & Definition
Hypocalcaemia is a serum total calcium below 2.1 millimoles per litre, or an ionised calcium below 1.1 millimoles per litre. Total calcium must be corrected for albumin: add 0.02 millimoles per litre for every gram per litre that albumin is below 40. The ionised calcium is the physiologically active fraction, and it is the better test in acutely ill children, neonates, and any child with acid–base disturbance, because albumin and protein binding shift with pH. [3] [8]
Hypoparathyroidism is the state in which parathyroid hormone is deficient (or, in pseudohypoparathyroidism, resisted), so calcium falls and phosphate rises. The clinical syndrome is neuromuscular irritability: paraesthesiae around the mouth and in the fingers, muscle cramping, carpopedal spasm, and in severe cases laryngospasm, bronchospasm, and generalised seizures. Infants and young children often present with a seizure as the first sign, with no preceding history of tingling. [1] [3]
Classification
The fastest way to classify hypocalcaemia is by the PTH level and the age of onset. The figure below splits causes into low-PTH (hypoparathyroid) and high-PTH (secondary) groups, and places neonatal hypocalcaemia in its age-specific band. [3] [8]
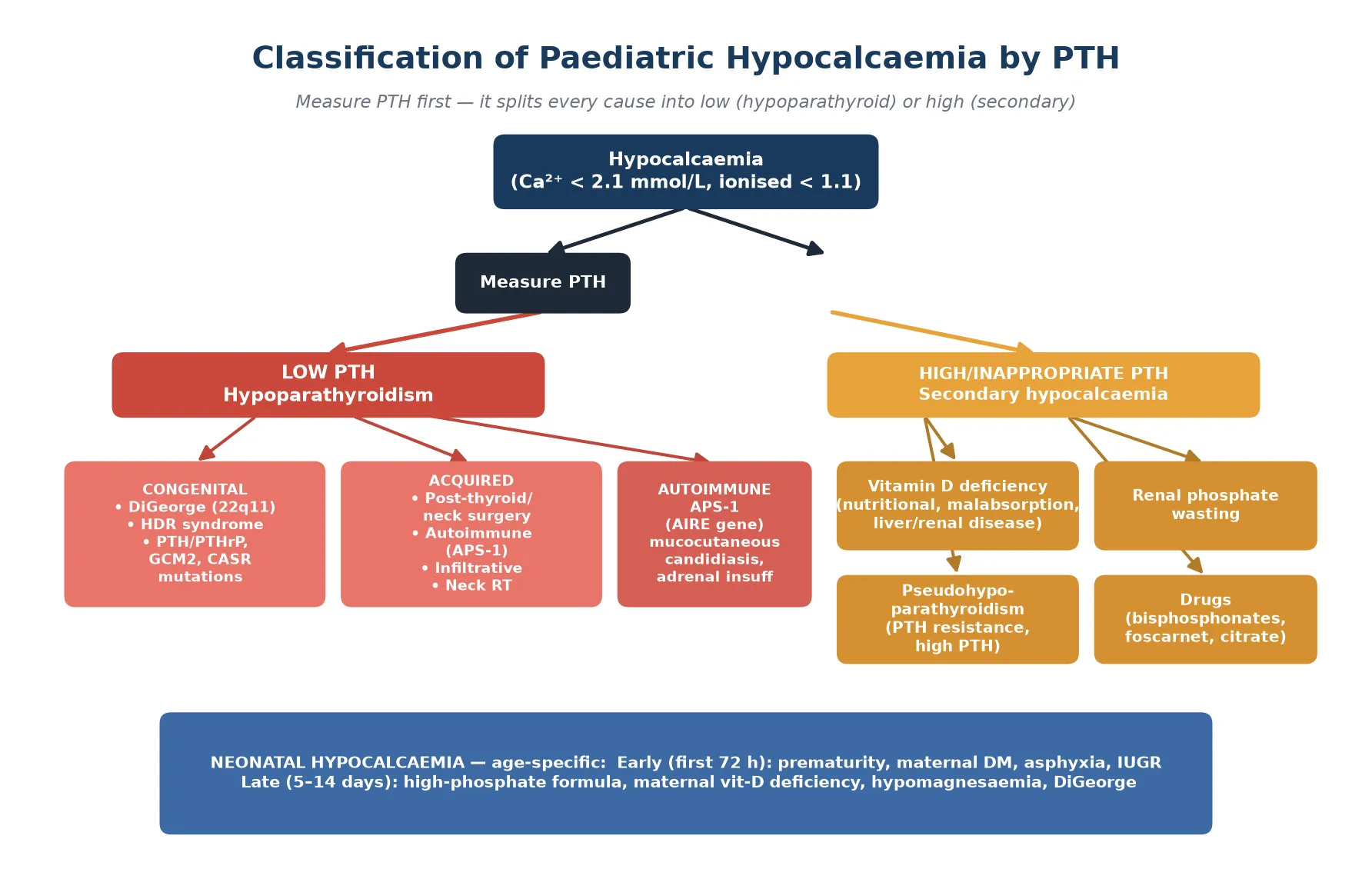
Hypoparathyroidism (low PTH)
- PTH deficient: gland damaged, absent, or functionally silenced
- Labs: ↓ Ca²⁺, ↑ PO₄³⁻, ↓ PTH, ↓ 1,25(OH)₂D
- Congenital: DiGeorge (22q11.2), HDR, GCM2, AIRE (APS-1) mutations
- Acquired: post-surgical, autoimmune, infiltrative, neck radiotherapy
Pseudohypoparathyroidism (high PTH)
- PTH present and high, but target organs resist it (GNAS mutation)
- Labs: ↓ Ca²⁺, ↑ PO₄³⁻, ↑↑ PTH — the PTH is high, not low
- Type 1a: Albright hereditary osteodystrophy phenotype (short stature, brachydactyly, obesity, round face)
- Treat with calcium + calcitriol; PTH analogue generally not first-line
Vitamin D deficiency (high PTH)
- Secondary hyperparathyroidism from inadequate gut calcium absorption
- Labs: ↓/normal Ca²⁺, ↓ PO₄³⁻, ↑ PTH, ↓ 25(OH)D (below 30 nmol/L deficient)
- May coexist with rickets on the skeletal exam
- Treat with cholecalciferol or ergocalciferol plus calcium
Neonatal hypocalcaemia
- Early (first 72 h): prematurity, maternal diabetes, birth asphyxia, IUGR
- Late (day 5–14): high-phosphate formula, maternal vitamin D deficiency, hypomagnesaemia, DiGeorge
- Ionised calcium is the reliable test in the neonate
- Most early cases resolve with correction of the trigger or formula change
Epidemiology & Risk Factors
Hypoparathyroidism is a rare endocrine disorder, with an estimated prevalence in adults of approximately 24 to 37 per 100,000; paediatric-onset disease is rarer still and is dominated by congenital and early-life causes. The single most common identifiable cause of hypoparathyroidism across all ages is anterior neck surgery — thyroidectomy or parathyroidectomy — but in children, congenital causes lead. [3] [7]
22q11.2 deletion syndrome (DiGeorge / velocardiofacial syndrome) is the most frequent congenital cause of hypoparathyroidism in childhood, with an incidence of approximately 1 in 4,000 live births. Hypocalcaemia develops in roughly 60 to 70 per cent of affected children, though it may be transient in infancy and recur later. A Finnish nationwide cohort confirmed that endocrine, cardiac, immunological, and neurodevelopmental manifestations cluster, and that hypocalcaemia is often the presenting feature that triggers the genetic diagnosis. [9]
Autoimmune polyglandular syndrome type 1 (APS-1), caused by AIRE gene mutations, is a rarer but high-yield exam cause of hypoparathyroidism. It classically presents the triad of chronic mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency, with hypoparathyroidism usually appearing first in early childhood. Pseudohypoparathyroidism type 1a is rarer still, with an estimated prevalence of less than 1 per 100,000, and is inherited maternally through GNAS imprinting. [3] [4]
CALCIUM
Facial nerve tap → ipsilateral facial twitch — a bedside sign of neuromuscular irritability (also seen in normal children, so not specific alone)
Correct total calcium for albumin: add 0.02 mmol/L per g/L below 40 — or use ionised calcium in the sick child
Low PTH + low Ca²⁺ = hypoparathyroidism; the gland is the problem
Calcium-sensing receptor — mutated in autosomal dominant hypocalcaemia and in the activating form that mimics hypoparathyroidism
The physiologically active fraction — the best test in neonates and acutely ill children
Monitor it in treated patients to avoid hypercalciuria and nephrocalcinosis — the treatment target is calcium just below normal
Severe hypomagnesaemia causes refractory hypocalcaemia by impairing PTH secretion AND action — check and replace it
Vitamin D deficiency is the most common cause of hypocalcaemia globally and the dominant high-PTH cause in children, particularly in breast-fed infants of vitamin-D-deficient mothers, dark-skinned children, those with limited sun exposure, and migrants and refugees from regions where supplementation is not routine. The global consensus on nutritional rickets sets the prevention standard: 400 international units of vitamin D daily from birth, with higher-risk groups requiring vigilance. [10]
Pathophysiology
Calcium homeostasis is maintained by a closed feedback loop involving parathyroid hormone, calcitriol (1,25-dihydroxyvitamin D), and — to a lesser extent — calcitonin. When ionised calcium falls, the calcium-sensing receptor on parathyroid chief cells triggers PTH release within seconds. PTH acts at three sites over three timescales: it mobilises calcium from bone within minutes, it increases renal calcium reabsorption and phosphate excretion within hours, and it stimulates renal 1-alpha-hydroxylase to produce calcitriol, which increases gut calcium absorption over days. The loop closes when rising calcium suppresses PTH release. [3] [8]
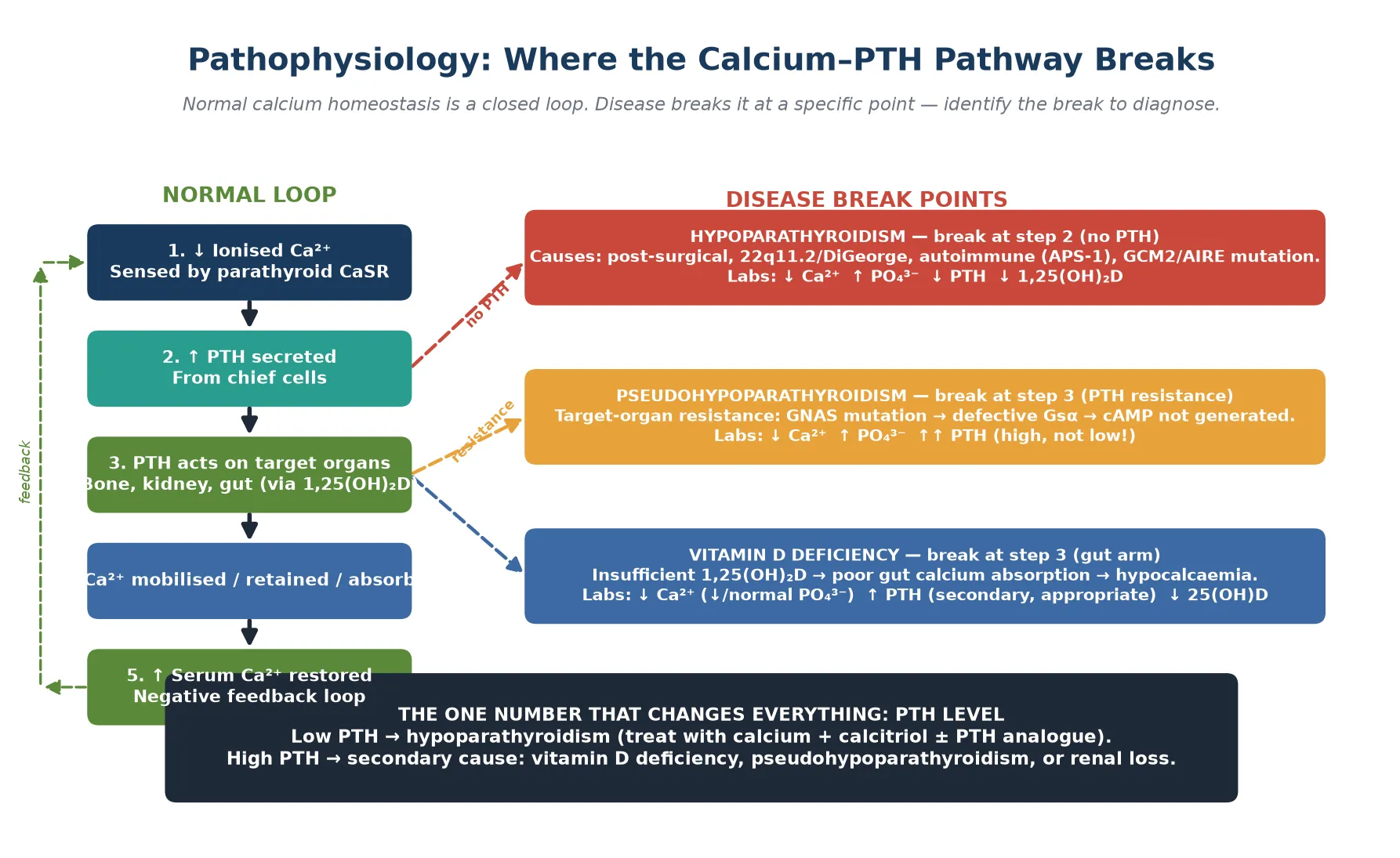
In true hypoparathyroidism, the parathyroid glands are absent, damaged, or functionally silenced. No PTH is released, so bone does not mobilise calcium, the kidney wastes calcium and retains phosphate, and calcitriol production falls. The result is low calcium, high phosphate, low PTH, and low calcitriol. The high phosphate is clinically important: it can precipitate as ectopic calcification and it lowers calcium further by mass action. In postsurgical hypoparathyroidism, the timeline is acute and the gland is physically removed or devascularised. In autoimmune disease (APS-1), autoantibodies or immune-mediated destruction silence the gland over time. [1] [3]
In pseudohypoparathyroidism, the problem is end-organ resistance. PTH is produced in abundance, but a mutation in GNAS — which encodes the Gs-alpha stimulatory protein that links the PTH receptor to cAMP generation — means the kidney and bone cannot respond. The biochemical signature is identical to hypoparathyroidism (low calcium, high phosphate) but with a high PTH. Type 1a carries the Albright hereditary osteodystrophy phenotype (short stature, round face, obesity, brachydactyly, subcutaneous ossifications); type 1b has the biochemical resistance without the skeletal phenotype; and pseudo-pseudohypoparathyroidism has the phenotype but normal biochemistry. [4]
In vitamin D deficiency, the gut arm of the loop fails. Inadequate 25-hydroxyvitamin D limits calcitriol production, gut calcium absorption falls, and the parathyroid gland responds appropriately with high PTH to maintain calcium at the cost of bone demineralisation. Calcium may be low or low-normal, phosphate is often low (because PTH drives phosphaturia), and 25-hydroxyvitamin D is deficient (below 30 nanomoles per litre). This is why measuring PTH and 25-hydroxyvitamin D together is the key to separating the high-PTH causes. [10]
Hypoparathyroidism
- PTH absent or low — gland damaged or absent
- Phosphate high (no PTH to excrete it)
- Calcitriol low (no PTH to drive 1-alpha-hydroxylase)
- Break at the source: parathyroid gland
Pseudohypoparathyroidism
- PTH high — gland works, target organs resist
- Phosphate high (kidney cannot respond to PTH-driven excretion)
- GNAS mutation → defective Gsα → no cAMP signal
- Break at the receptor: end-organ resistance
Clinical Presentation
The presentation of hypocalcaemia is the presentation of neuromuscular irritability. The child feels tingling around the mouth and in the fingers and toes, develops muscle cramps, and may progress to carpopedal spasm — the hand forced into the accoucheur position with metacarpophalangeal flexion and interphalangeal extension. Severe hypocalcaemia causes laryngospasm (stridor), bronchospasm, and generalised tonic-clonic seizures. Infants and young children often present with a seizure as the first sign, with no warning history of tingling, because they cannot describe paraesthesiae. [1] [3]
Two bedside signs support the diagnosis of latent tetany when the calcium is borderline. Chvostek's sign is twitching of the ipsilateral facial muscles on tapping the facial nerve just anterior to the ear. Trousseau's sign is carpopedal spasm induced by inflating a blood pressure cuff above systolic pressure for three minutes. Trousseau's is the more specific of the two; Chvostek's is present in up to 10 per cent of normal children, so it should never be relied on alone. Both are clinical adjuncts — the ionised calcium is the diagnostic test. [3]
A neonate with hypocalcaemia may present with jitteriness, irritability, poor feeding, apnoea, or frank seizures. Early neonatal hypocalcaemia (first 72 hours) is common in premature infants, infants of diabetic mothers, and infants with perinatal asphyxia, because the parathyroid axis is immature and the calcium flux across the placenta has ceased. Late neonatal hypocalcaemia (day 5 to 14) points to high-phosphate formula feeding, maternal vitamin D deficiency, hypomagnesaemia, or DiGeorge syndrome. The infant with a heart murmur, hypocalcaemia, and absent thymic shadow on chest imaging has 22q11.2 deletion until proven otherwise. [8] [9]
The child with chronic hypoparathyroidism may also show features of the underlying syndrome. Look for the facial features of DiGeorge (small chin, low-set ears, hooded eyelids, cleft palate), the skeletal phenotype of pseudohypoparathyroidism type 1a (short stature, round face, brachydactyly, short fourth metacarpal), or the mucocutaneous candidiasis and skin and nail dystrophy of APS-1. A child who develops hypocalcaemia in the days after thyroid or neck surgery has postsurgical hypoparathyroidism until proven otherwise. [1] [4]
Differential Diagnosis
The differential of a low calcium is organised by the PTH level and the clinical context. A low PTH points to hypoparathyroidism — congenital (DiGeorge, HDR syndrome, isolated GCM2 or AIRE mutations, activating CASR mutation) or acquired (postsurgical, autoimmune, infiltrative, neck radiotherapy). A high PTH points to a secondary cause: vitamin D deficiency, pseudohypoparathyroidism, renal phosphate wasting, malabsorption, or acute severe illness with hypocalcaemia. [3] [7]
The single most important discriminating test is the PTH measured with the calcium. A low PTH with low calcium is hypoparathyroidism regardless of age. A high PTH with low calcium is an appropriate gland response to a peripheral problem, and the next test is 25-hydroxyvitamin D to separate vitamin D deficiency from pseudohypoparathyroidism. Phosphate helps: a high phosphate favours hypoparathyroidism or pseudohypoparathyroidism (no renal phosphate excretion); a low or normal phosphate favours vitamin D deficiency (PTH-driven phosphaturia). [3]
Points to hypoparathyroidism
- Low Ca²⁺ with low PTH
- High phosphate (no PTH-driven excretion)
- Post-neck-surgery, or congenital syndrome features
- Low or low-normal calcitriol
Points to vitamin D deficiency
- Low/normal Ca²⁺ with high PTH
- Low phosphate (PTH-driven phosphaturia)
- Low 25(OH)D (below 30 nmol/L deficient)
- Breast-fed infant, dark skin, limited sun, rickets signs
Points to pseudohypoparathyroidism
- Low Ca²⁺ with HIGH PTH and high phosphate
- Albright hereditary osteodystrophy phenotype (type 1a)
- GNAS mutation, maternal inheritance pattern
- Elevated PTH confirms resistance, not deficiency
A bedside pearl: if the calcium will not rise despite adequate calcium and calcitriol, check the magnesium. Hypomagnesaemia is the great mimic and the great refractory cause, and it is the single most commonly missed reversible factor in hypocalcaemia that "is not responding." Also exclude hyperphosphataemia as a driver — phosphate binds calcium, and a child in tumour lysis or rhabdomyolysis can be hypocalcaemic purely from phosphate binding. [3] [7]
Clinical & Bedside Assessment
The assessment of a child with suspected hypocalcaemia begins with the airway, breathing, and circulation, because severe hypocalcaemia causes laryngospasm, bronchospasm, and seizures. Establish whether the child is symptomatic right now: a seizing child, a child with stridor, or a child with carpopedal spasm needs intravenous calcium immediately. Only an asymptomatic child with an incidental low calcium should wait for the full panel before treatment. [1] [3]
Look for the bedside signs of latent tetany. Test Chvostek's sign by tapping the facial nerve anterior to the ear and watching for ipsilateral facial muscle twitching. Test Trousseau's sign by inflating a blood pressure cuff above systolic pressure for three minutes and watching for carpopedal spasm. Examine for the phenotypic features of the underlying syndrome: the facial features of DiGeorge, the skeletal phenotype of pseudohypoparathyroidism, the candidiasis and ectodermal dystrophy of APS-1, and the surgical scar of a recent thyroidectomy. [4] [9]
Take a targeted history. Ask about the timeline of symptoms, recent surgery (thyroidectomy, parathyroidectomy, neck dissection), family history of calcium or endocrine disorders, maternal vitamin D status and supplementation, feeding history (formula phosphate content), and the developmental and cardiac history that points to 22q11.2 deletion. In an adolescent girl with hypocalcaemia and a recent thyroid operation, the diagnosis is postsurgical hypoparathyroidism until serial calcium confirms recovery or permanence. [8] [9]
Investigations
The investigation of hypocalcaemia runs in tiers: an acute bundle that confirms severity and screens for the emergency, then a PTH-led classification panel, then genetic and imaging tests for the underlying cause. [3] [7]
Investigation bundle for hypocalcaemia
Ionised calcium (or total calcium corrected for albumin) — confirms hypocalcaemia and severity
PTH — the single most important test: low = hypoparathyroidism, high = secondary cause
Phosphate — high favours hypoparathyroidism or pseudohypoparathyroidism; low favours vitamin D deficiency
Magnesium — check in every refractory case; severe hypomagnesaemia causes PTH failure
25-hydroxyvitamin D — separates vitamin D deficiency from pseudohypoparathyroidism in the high-PTH group
Renal function and urinary calcium-to-creatinine ratio — baseline for treatment monitoring
ECG — look for prolonged QT interval, a marker of cardiac irritability and seizure risk
If congenital suspected: 22q11.2 deletion FISH or microarray; cardiac echo; immune panel
If autoimmune suspected: anti-NALP5 and anti-cytokine antibodies, cortisol, ACTH (APS-1 screen)
The ECG is part of the acute assessment because hypocalcaemia prolongs the QT interval and increases the risk of arrhythmia and seizure. A corrected QT interval above 470 milliseconds in a child with hypocalcaemia warrants urgent correction and cardiac monitoring. Correct the calcium slowly, because rapid intravenous calcium can cause bradycardia and shortening of the QT in the other direction. [1]
Long-term PTH 1-34 replacement in children (Winer 2018)
J Pediatr
Open-label study of long-term synthetic PTH 1-34 replacement in children with hypoparathyroidism, comparing once-daily and twice-daily dosing against conventional calcium and calcitriol therapy, with serum and urinary calcium as primary endpoints.
Key finding
PTH 1-34 maintained serum calcium in the target range with reduced calcium and calcitriol supplementation requirements, and twice-daily dosing produced more stable calcium levels than once-daily. Urinary calcium excretion was lower than with conventional therapy, suggesting a renal calcium-sparing benefit.
Practice change
PTH 1-34 is a viable long-term option for selected children with refractory hypoparathyroidism, reducing the calcium and calcitriol burden and the risk of hypercalciuria, though it requires specialist endocrine oversight and careful monitoring.
Genetic testing is indicated when a congenital cause is suspected: 22q11.2 deletion testing (FISH or chromosomal microarray) for DiGeorge, GNAS methylation and sequencing for pseudohypoparathyroidism, AIRE sequencing for APS-1, and CASR, GCM2, or GATA3 sequencing for the rarer familial forms. A cardiac echo is essential in any child with suspected 22q11.2 deletion, because conotruncal heart defects (tetralogy of Fallot, interrupted aortic arch, truncus arteriosus) are common and life-threatening. [9]
Management — Resuscitation
Acute symptomatic hypocalcaemia — seizure, carpopedal spasm, laryngospasm, or stridor — is a medical emergency. The treatment is intravenous 10 per cent calcium gluconate at 0.5 millilitres per kilogram (maximum 20 millilitres) given slowly over 5 to 10 minutes with continuous cardiac monitoring. Calcium gluconate is preferred over calcium chloride for peripheral venous access because it is less vesicant if extravasation occurs. Never give intravenous calcium rapidly: it causes bradycardia, arrhythmia, and can arrest the heart in systole. [1] [3]
After the bolus, start a continuous intravenous calcium infusion if the child remains symptomatic or the calcium is critically low, then transition to oral calcium and calcitriol once stable. The infusion dose is 10 to 20 milligrams per kilogram per hour of elemental calcium, titrated to the serum level and the symptoms. Check the magnesium in every case: if it is low, replace it, because no amount of calcium will correct hypocalcaemia driven by hypomagnesaemia. [3]
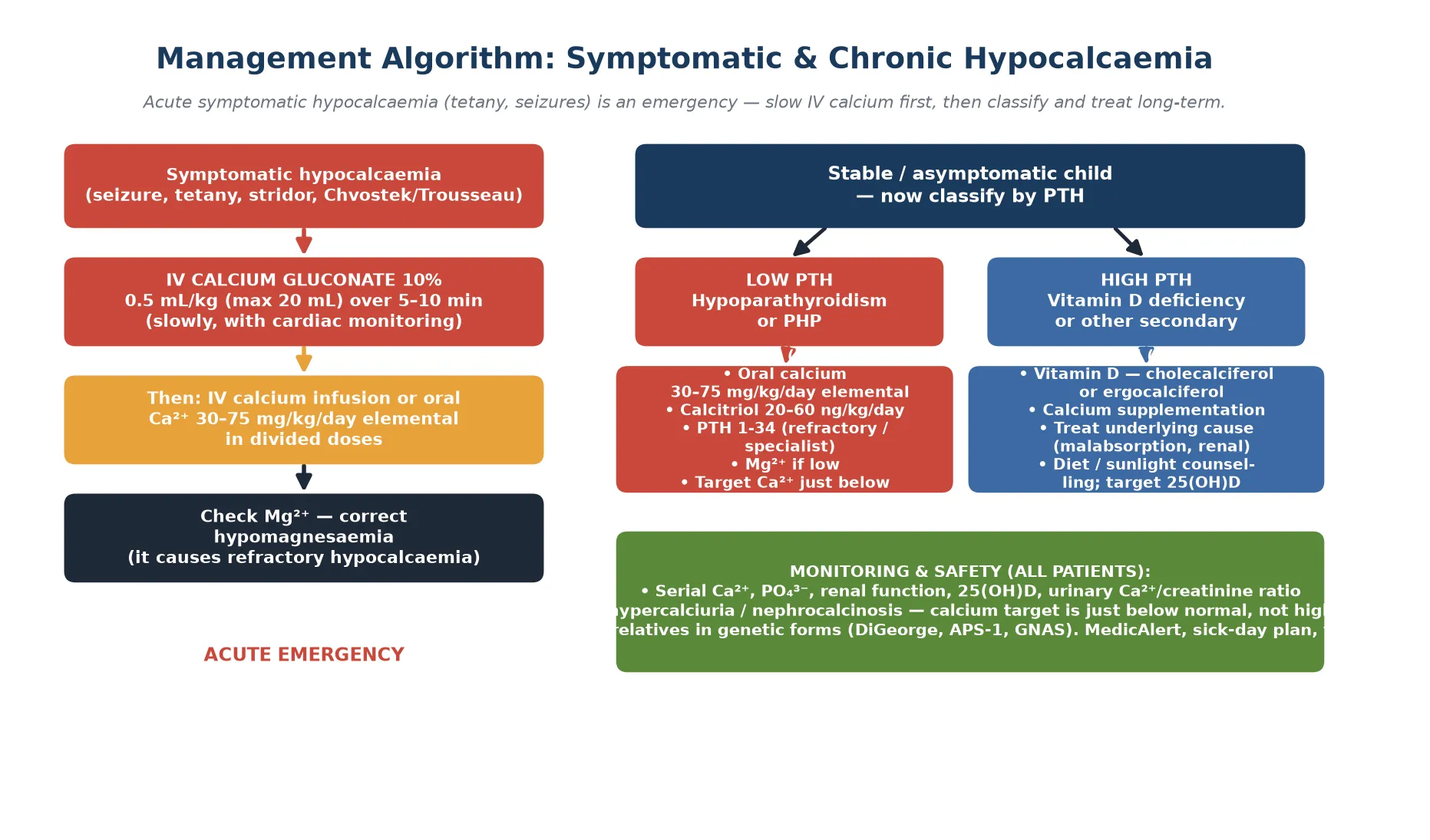
For the child with an incidental, asymptomatic low calcium, oral calcium alone may suffice while the cause is investigated. Oral elemental calcium at 30 to 75 milligrams per kilogram per day in divided doses raises the serum calcium over hours to days. Start calcitriol (20 to 60 nanograms per kilogram per day) when hypoparathyroidism or pseudohypoparathyroidism is confirmed, because native vitamin D cannot be activated without PTH. [1] [7]
Across the international guidelines (Brandi 2016, Khan 2022, Khan 2025), acute symptomatic hypocalcaemia is managed identically: slow intravenous calcium gluconate with cardiac monitoring, followed by oral calcium and active vitamin D. The guidelines converge on treating to a target calcium just below the normal range — not high-normal — to minimise hypercalciuria and nephrocalcinosis, which are the principal long-term harms of overtreatment. [1] [7]
Management — Definitive & Stepwise
The definitive management of hypoparathyroidism rests on a pair of agents: oral calcium and calcitriol (1,25-dihydroxyvitamin D). Oral elemental calcium is given at 30 to 75 milligrams per kilogram per day in divided doses, titrated to the serum calcium. Calcitriol is given at 20 to 60 nanograms per kilogram per day, because native vitamin D cannot be converted to its active form without PTH. The combination raises calcium while controlling phosphate, and the dose is titrated to keep the serum calcium just below the normal range. [1] [2]
Calcium and calcitriol (standard hypoparathyroidism regimen)
For refractory hypoparathyroidism — where calcium and calcitriol cannot maintain safe levels without causing hypercalciuria — PTH 1-34 (teriparatide or native synthetic PTH 1-34) is an option under specialist paediatric endocrine care. The Winer studies demonstrated that long-term PTH 1-34 in children maintains calcium in target with lower supplemental calcium and calcitriol doses and reduced urinary calcium, and that twice-daily dosing produces more stable levels than once-daily. PTH 1-84 is approved for adults but data in children are limited; PTH 1-34 remains the paediatric evidence base. [5] [6]
Definitive pathway for confirmed hypoparathyroidism
Confirm diagnosis: low Ca²⁺ with low PTH, high phosphate, normal renal function
Start oral elemental calcium 30–75 mg/kg/day in divided doses
Add calcitriol 20–60 ng/kg/day (native vitamin D will not work)
Check and replace magnesium if low — it is the great refractory cause
Titrate to serum Ca²⁺ just below normal — not high-normal — to protect the kidney
Monitor: serum Ca²⁺, PO₄³⁻, renal function, urinary Ca²⁺/creatinine ratio
If refractory or hypercalciuria: refer for PTH 1-34 under specialist care
Identify and manage the underlying cause (genetic testing, immune screen)
MedicAlert identifier, sick-day plan, and structured transition to adult endocrine care
For pseudohypoparathyroidism, the management is calcium and calcitriol — the same as for hypoparathyroidism — because the goal is to correct the hypocalcaemia, not to supply PTH (which the body is already making in excess but cannot use). The Mantovani consensus standardised the diagnosis and subtype classification, which guides genetic counselling and surveillance for the associated endocrine and developmental features. For vitamin D deficiency, the treatment is cholecalciferol or ergocalciferol with calcium, and the prevention standard is 400 international units of vitamin D daily from birth. [4] [10]
Specific Subtypes & Scenarios
The four scenarios below are the ones that present acutely, confuse the team, and appear in exams. [3] [8]
Neonatal hypocalcaemia is classified by timing. Early hypocalcaemia (first 72 hours) is driven by an immature parathyroid axis, a sudden cessation of placental calcium supply, and prematurity or perinatal stress (asphyxia, maternal diabetes, intrauterine growth restriction). It is usually asymptomatic and self-limiting, resolving as the axis matures. Late hypocalcaemia (day 5 to 14) is driven by a high-phosphate load (cows-milk-based formula), maternal vitamin D deficiency, hypomagnesaemia, or DiGeorge syndrome. Symptomatic late hypocalcaemia with seizures warrants a full work-up including 22q11.2 testing. [8]
DiGeorge syndrome (22q11.2 deletion) is the archetype of syndromic hypoparathyroidism. The child presents with hypocalcaemia (often transient in infancy), conotruncal heart disease, thymic aplasia with immune deficiency, cleft palate, and characteristic facial features. The hypocalcaemia may be the presenting feature that triggers the genetic diagnosis, and it can recur years after a quiescent period. Every child with unexplained hypocalcaemia and a heart murmur or immune deficiency needs 22q11.2 testing. [9]
Postsurgical hypoparathyroidism follows thyroidectomy, parathyroidectomy, or radical neck dissection. The hypocalcaemia develops within 24 to 72 hours as the glands are removed, devascularised, or stunned. Most cases are transient, recovering over weeks, but some are permanent. The countermeasure is serial calcium every 12 hours for the first 48 hours, early oral calcium and calcitriol, and intravenous calcium for symptomatic dips. If the PTH is undetectable immediately post-operatively, the risk of permanence is higher. [1] [7]
Autoimmune polyglandular syndrome type 1 (APS-1) presents with the classic triad of chronic mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency, caused by AIRE gene mutations. Hypoparathyroidism usually appears first, in early childhood. The adrenal insufficiency may follow years later, so every child with APS-1 needs lifelong cortisol surveillance. The candidiasis is often the earliest visible clue — chronic thrush that does not respond to standard treatment. [3]
Complications & Pitfalls
The complications of hypocalcaemia are the complications of the disease (seizures, laryngospasm, cardiac arrhythmia from prolonged QT, and — in chronic untreated disease — intracranial calcification and cataracts) and the complications of its treatment (hypercalciuria, nephrocalcinosis, and nephrolithiasis from over-treatment). The balance of therapy is to raise calcium enough to prevent symptoms without driving it so high that the kidney suffers. [1] [3]
Disease pitfalls
- Missing a seizing child's hypocalcaemia because the total calcium looks normal (albumin correction or ionised needed)
- Treating refractory hypocalcaemia without checking magnesium
- Missing DiGeorge in an infant with hypocalcaemia and a heart murmur
- Labeling high-PTH hypocalcaemia as hypoparathyroidism (it is secondary, not primary)
- Missing adrenal insufficiency in APS-1 (hypoparathyroidism comes first)
Treatment pitfalls
- Giving intravenous calcium too fast (bradycardia, cardiac arrest in systole)
- Extravasation of calcium chloride through a peripheral line (tissue necrosis)
- Over-treating to a high-normal calcium (hypercalciuria, nephrocalcinosis)
- Using native vitamin D instead of calcitriol in hypoparathyroidism (it cannot be activated)
- Not monitoring urinary calcium in treated patients
The most dangerous acute pitfall is giving intravenous calcium too rapidly. Calcium gluconate given as a fast push causes bradycardia, arrhythmia, and can arrest the heart in systole. The countermeasure is to give it slowly over 5 to 10 minutes with continuous cardiac monitoring, and to stop if the heart rate drops. The second acute pitfall is peripheral extravasation: calcium chloride is highly vesicant and causes tissue necrosis, so it is reserved for central access, while calcium gluconate is the peripheral agent. [1] [3]
The most common chronic pitfall is over-treatment. Driving the calcium to the high-normal range — which feels satisfying — causes hypercalciuria (because there is no PTH to reabsorb calcium in the kidney), nephrocalcinosis, and nephrolithiasis over months to years. The treatment target is a calcium just below the normal range, sufficient to prevent symptoms and seizures but low enough to protect the kidney. The urinary calcium-to-creatinine ratio is the surveillance tool for this. [2] [7]
Prognosis & Disposition
The prognosis of hypoparathyroidism depends on the cause and the quality of long-term management. Postsurgical hypoparathyroidism is often transient, recovering over weeks to months as stunned glands recover; the remainder are permanent. Congenital and genetic forms (DiGeorge, APS-1, pseudohypoparathyroidism, familial mutations) are lifelong, but with careful calcium and calcitriol management — and PTH 1-34 where indicated — growth, development, and quality of life are good. [1] [3]
The prognosis of vitamin D deficiency hypocalcaemia is excellent with adequate supplementation and dietary correction. The prognosis of pseudohypoparathyroidism depends on the subtype and the management of associated endocrine and developmental features; the skeletal phenotype of type 1a is present from childhood, and the biochemical resistance is lifelong. [4] [10]
Disposition is lifelong for the permanent forms. The child leaves the acute admission on oral calcium and calcitriol (with magnesium replaced if deficient), with a MedicAlert identifier, a sick-day plan (because illness disrupts intake and absorption), and a named paediatric endocrinologist. The family is counselled on the genetic basis where relevant, and screening of at-risk relatives is arranged in hereditary forms. Structured transition to adult endocrinology in late adolescence carries the dosing, the monitoring, and the genetic counselling forward. [1] [7]
Special Populations
The infant with DiGeorge syndrome carries the highest burden of associated disease. The hypocalcaemia is often transient in the neonatal period but can recur years later, so calcium surveillance must continue even after apparent resolution. The cardiac anatomy dictates the early prognosis (conotruncal defects), the thymic aplasia dictates the immune surveillance, and the neurodevelopmental phenotype (learning difficulty, psychiatric illness in adolescence) requires longitudinal developmental support. The endocrine, cardiac, immunology, and developmental teams must coordinate. [9]
The child with APS-1 faces lifelong multi-endocrine risk. Hypoparathyroidism is usually first, but adrenal insufficiency follows and is life-threatening if missed. Every APS-1 child needs serial morning cortisol and ACTH stimulation testing, because adrenal crisis can be the first sign of the second deficiency. The chronic mucocutaneous candidiasis is chronic and resistant, and ectodermal dystrophy (enamel hypoplasia, nail dystrophy, keratoconjunctivitis) is part of the syndrome. [3]
The refugee, migrant, or socioeconomically disadvantaged child is at highest risk of vitamin D deficiency hypocalcaemia, because maternal deficiency, exclusive breastfeeding without supplementation, dark skin, limited sun exposure, and cultural dress practices converge. The prevention standard — 400 international units of vitamin D daily from birth — is the single most cost-effective intervention, and screening at-risk groups on arrival is part of equitable paediatric care. Rickets may be the presenting feature, and the hypocalcaemia can be severe enough to seize. [10]
The child with complex chronic illness or technology dependence (chronic kidney disease, malabsorption, short-gut syndrome, on loop diuretics or bisphosphonates) has hypocalcaemia from multiple converging mechanisms, and the management must address each layer. These children need a coordinated endocrine, renal, and nutrition team, and close calcium surveillance during illness or medication changes. [2] [7]
Evidence, Guidelines & Regional Differences
The evidence base for paediatric hypoparathyroidism rests on three international consensus statements (Brandi 2016, Khan 2022, Khan 2025), the Nature Reviews Disease Primers overview (Mannstadt 2017), the pseudohypoparathyroidism consensus (Mantovani 2018), the Winer PTH 1-34 paediatric trials, and the perinatal calcium physiology review (Hsu and Levine 2004). The consensus statements set the modern standard for diagnosis, monitoring targets, and the role of PTH analogues, and they converge on treating to a calcium just below normal to protect the kidney. [1] [2] [7]
The Winer studies established PTH 1-34 as a viable long-term option in children, demonstrating calcium stability with lower urinary calcium than conventional therapy. This evidence underpins the shift toward hormone replacement — rather than just calcium and calcitriol supplementation — in refractory paediatric hypoparathyroidism, though access to PTH 1-34 for children varies by jurisdiction and reimbursement. [5] [6]
Australia and Aotearoa New Zealand manage paediatric hypoparathyroidism through tertiary paediatric endocrinology services, with acute symptomatic hypocalcaemia managed in regional and tertiary hospitals using intravenous calcium gluconate. Calcitriol is the standard active vitamin D, and PTH 1-34 is available through specialist centres for refractory disease. Vitamin D deficiency prevention follows the global consensus standard of 400 international units daily from birth, with targeted screening of at-risk groups including refugees, dark-skinned children, and exclusively breastfed infants. The Australasian Paediatric Endocrine Group supports clinical guidance and transition. [1] [10]
The remaining controversies are practical: the optimal PTH 1-34 formulation and dosing schedule for children, the long-term safety of continuous PTH analogue exposure (theoretical osteosarcoma signal from rodent studies, not seen in human follow-up), and the threshold for genetic testing in apparently isolated hypoparathyroidism. These are genuinely evolving, and a fellowship candidate names the direction of travel rather than pretending the field is settled. [5] [7]
Exam Pearls
One-sentence answer: the approach to a hypocalcaemic child
A child with hypocalcaemia is managed by treating symptomatic disease first (intravenous calcium gluconate 0.5 mL per kg slowly with cardiac monitoring), then classifying by PTH (low PTH is hypoparathyroidism, treated with oral calcium and calcitriol with PTH 1-34 for refractory disease; high PTH is vitamin D deficiency or pseudohypoparathyroidism, treated with vitamin D and calcium), checking magnesium in every refractory case, and targeting a calcium just below normal to avoid hypercalciuria and nephrocalcinosis.
Definition and the PTH split
- Hypocalcaemia: total Ca²⁺ < 2.1 mmol/L or ionised < 1.1 mmol/L (correct for albumin)
- PTH is the first test: low = hypoparathyroidism; high = vitamin D deficiency or pseudohypoparathyroidism
- Phosphate helps: high PO₄³⁻ favours hypopara/PHP; low favours vitamin D deficiency
Acute emergency management
- Symptomatic (seizure, tetany, stridor): IV 10% calcium gluconate 0.5 mL/kg (max 20 mL) over 5–10 min
- Give SLOWLY with cardiac monitoring — rapid push causes bradycardia and arrest
- Calcium gluconate for peripheral lines; calcium chloride only for central access (vesicant)
- Always check and replace magnesium in refractory hypocalcaemia
Chronic hypoparathyroidism
- Oral elemental calcium 30–75 mg/kg/day + calcitriol 20–60 ng/kg/day
- Native vitamin D does NOT work (no PTH to activate it) — use calcitriol
- Target Ca²⁺ just below normal to avoid hypercalciuria and nephrocalcinosis
- Refractory: PTH 1-34 under specialist care (Winer evidence)
Syndromic causes
- DiGeorge (22q11.2): hypocalcaemia + heart defect + thymic aplasia + cleft palate
- APS-1 (AIRE): hypoparathyroidism + mucocutaneous candidiasis + adrenal insufficiency
- Pseudohypoparathyroidism 1a: low Ca²⁺, high PTH, Albright hereditary osteodystrophy phenotype
- Neonatal late hypocalcaemia (day 5–14): high-phosphate formula, maternal vit-D deficiency, DiGeorge
Frequently misremembered facts, stated correctly: pseudohypoparathyroidism has a high PTH, not a low one — the gland works, the target organs resist. Chvostek's sign is present in up to 10 per cent of normal children, so it does not diagnose hypocalcaemia alone. Native vitamin D (cholecalciferol) cannot treat hypoparathyroidism, because there is no PTH to drive the renal 1-alpha-hydroxylation step — calcitriol is required. And the treatment target in chronic hypoparathyroidism is a calcium just below normal, not high-normal, because the unprotected kidney develops hypercalciuria, nephrocalcinosis, and stones. [1] [4]
The one-diagram-that-answers-everything for a fellowship exam is the calcium-PTH axis: calcium falls, parathyroid releases PTH, PTH acts on bone and kidney and gut, calcium is restored. Every question on hypocalcaemia maps onto this pathway. Hypoparathyroidism breaks it before the PTH. Pseudohypoparathyroidism breaks it at the receptor. Vitamin D deficiency breaks it at the gut. Know where the break is, and the investigation and treatment follow. [3] [8]
References
- [1]Brandi ML; Bilezikian JP; Shoback D; et al Management of Hypoparathyroidism: Summary Statement and Guidelines. J Clin Endocrinol Metab, 2016.PMID 26943719
- [2]Khan AA; Koch CA; Van Uum SHM; et al Evaluation and Management of Hypoparathyroidism Summary Statement and Guidelines from the Second International Workshop. J Bone Miner Res, 2022.PMID 36054621
- [3]Mannstadt M; Bilezikian JP; Thakker RV; et al Hypoparathyroidism. Nat Rev Dis Primers, 2017.PMID 28857066
- [4]Mantovani G; Bastepe M; Monk D; et al Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol, 2018.PMID 29959430
- [5]Winer KK; Kelly A; Johns A; et al Long-Term Parathyroid Hormone 1-34 Replacement Therapy in Children with Hypoparathyroidism. J Pediatr, 2018.PMID 30470382
- [6]Winer KK; Sinaii N; Peterson D; et al Effects of once versus twice-daily parathyroid hormone 1-34 therapy in children with hypoparathyroidism. J Clin Endocrinol Metab, 2008.PMID 18492754
- [7]Khan AA; Clarke BL; Rejnmark L; et al Best practice recommendations for the diagnosis and management of hypoparathyroidism. Metabolism, 2025.PMID 40581321
- [8]Hsu SC; Levine MA Perinatal calcium metabolism: physiology and pathophysiology. Semin Neonatol, 2004.PMID 15013473
- [9]Wahrmann S; Jokinen E; Pitkänen S; et al Childhood manifestations of 22q11.2 deletion syndrome: A Finnish nationwide register-based cohort study. Acta Paediatr, 2023.PMID 36867048
- [10]Munns CF; Shaw N; Kiely M; et al Global Consensus Recommendations on Prevention and Management of Nutritional Rickets. J Clin Endocrinol Metab, 2016.PMID 26745253