Paeds · endocrinology-diabetes-and-growth
Hypopituitarism and pituitary lesions
Also known as Hypopituitarism · Combined pituitary hormone deficiency · Panhypopituitarism · Congenital hypopituitarism · Septo-optic dysplasia · Pituitary stalk interruption syndrome · Craniopharyngioma · Intracranial germinoma · Central hypothyroidism · Central adrenal insufficiency
Fellowship guide to hypopituitarism and pituitary and hypothalamic lesions in children: the congenital forms (transcription-factor defects, septo-optic dysplasia, pituitary stalk interruption) and the acquired lesions (craniopharyngioma and germinoma), the anterior and posterior hormone deficiencies and the order in which they fail, the dynamic tests that confirm each axis, and the single replacement rule that keeps a child alive — glucocorticoid before thyroxine.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single idea that organises this topic is that a pituitary problem is really two problems stacked together: a hormone-deficiency problem and, when a lesion is the cause, a mass problem. The deficiency problem is solved axis by axis with paired pituitary-and-target hormones and dynamic tests, and the mass problem is solved by imaging the hypothalamic–pituitary region and treating the lesion. Miss either half and the child comes to harm. [1] [5]
This page covers congenital and acquired hypopituitarism, the pituitary and hypothalamic lesions that cause it in children — craniopharyngioma and germinoma foremost — the anterior and posterior hormone deficiencies, the dynamic tests that confirm each axis, and the replacement strategy. It links to the dedicated diabetes-insipidus and adrenal-insufficiency leaves for their full pathways rather than repeating them. [11] [9]
Overview & Definition
Hypopituitarism means the pituitary gland is not delivering enough of one or more of its hormones. When several anterior hormones fail together the term is combined pituitary hormone deficiency, and when all of them fail it is panhypopituitarism. The deficiency can be of the anterior lobe, the posterior lobe, or both, and it can arise at the level of the hypothalamus, the stalk, or the gland itself. [1]
The distinction that matters most clinically is congenital versus acquired. A congenital cause is present from birth — a mutation in a pituitary transcription factor, a midline malformation such as septo-optic dysplasia, or an interrupted pituitary stalk — and it declares itself in the newborn with hypoglycaemia and prolonged jaundice, or later with short stature and failure to progress through puberty. An acquired cause develops in a previously well child, and in childhood the commonest acquired cause is a tumour of the hypothalamic–pituitary region. [2] [5]
Because the pituitary sits beneath the optic chiasm and beside the hypothalamic centres for appetite, temperature and thirst, a lesion here rarely causes hormone failure alone. Visual field loss, an obesity or growth-arrest syndrome, disturbed sleep and central diabetes insipidus travel alongside the endocrine picture, which is why the evaluation is always both endocrine and neurological. [1] [5]
Classification
Sort the disease first by timing — congenital or acquired — because that split predicts the cause, the associated malformations and the surveillance a child needs. The figure below lays the congenital genetic and structural causes against the acquired tumoral, treatment-related and infiltrative causes, and separates anterior from posterior deficiencies. [2]
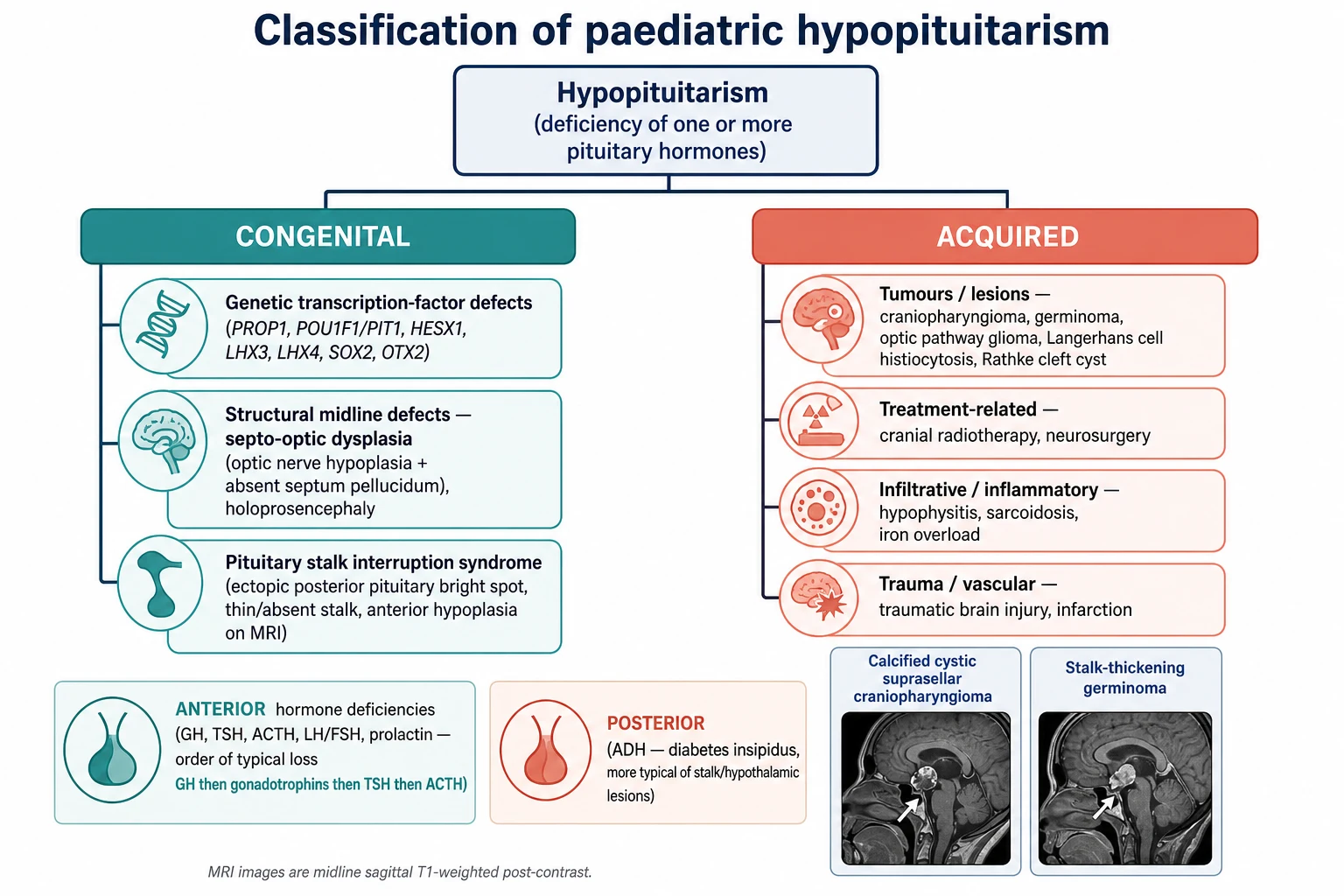
Congenital hypopituitarism
- Transcription-factor defects: PROP1, POU1F1, HESX1, LHX3, LHX4, SOX2, OTX2
- Midline malformation: septo-optic dysplasia, holoprosencephaly
- Pituitary stalk interruption syndrome on MRI
- Presents as neonatal hypoglycaemia or later short stature
Acquired lesions
- Craniopharyngioma: cystic, calcified, suprasellar
- Germinoma: midline, marker-secreting, stalk-thickening
- Optic pathway glioma, Langerhans cell histiocytosis, Rathke cleft cyst
- Cranial radiotherapy and neurosurgery as iatrogenic causes
By axis affected
- Anterior: growth hormone, LH and FSH, thyroid-stimulating hormone, adrenocorticotrophin, prolactin
- Posterior: antidiuretic hormone (central diabetes insipidus)
- Order of anterior loss: growth hormone earliest, adrenocorticotrophin latest
- Posterior failure flags a hypothalamic or stalk lesion
The genetic causes track the developmental hierarchy of the gland. Early transcription factors such as HESX1 govern the whole forebrain and pituitary, so their loss gives septo-optic dysplasia with wide deficiencies, whereas later factors such as PROP1 and POU1F1 govern specific cell lineages, so their loss gives a narrower, evolving pattern that classically spares adrenocorticotrophin at first. Knowing where a factor sits in the cascade predicts which hormones will fail and when. [2] [4]
Epidemiology & Risk Factors
Congenital hypopituitarism is rare, on the order of one in several thousand births, but it is under-recognised because the newborn signs are non-specific and the growth signs take months to declare. Isolated growth hormone deficiency is the commonest single anterior deficiency, and a proportion of children who begin with isolated deficiency evolve to combined deficiency over years, which is why one normal axis today does not close the file. [4] [8]
Craniopharyngioma is the commonest tumour to cause acquired hypopituitarism in children, with a peak incidence between five and fourteen years, and it accounts for a substantial share of childhood sellar and suprasellar tumours. Intracranial germinoma is less common but disproportionately important because it is highly treatable and because it can present with diabetes insipidus and stalk thickening months to years before a mass is visible. [5] [6]
The risk factors for acquired hypopituitarism are the lesions themselves and their treatment. Cranial radiotherapy for brain tumours or leukaemia damages the hypothalamic–pituitary axis in a dose- and time-dependent way, with growth hormone the most radiosensitive axis, so any irradiated child needs lifelong endocrine surveillance. Traumatic brain injury, neurosurgery and infiltrative disease such as Langerhans cell histiocytosis complete the acquired list. [1] [5]
Pathophysiology
To see why pituitary lesions cause the deficiencies they do, follow the traffic through the stalk. The hypothalamus makes releasing hormones — growth-hormone-releasing hormone, thyrotrophin-releasing hormone, corticotrophin-releasing hormone and gonadotrophin-releasing hormone — and sends them down the portal vessels of the stalk to the anterior pituitary, while antidiuretic hormone and oxytocin travel down axons of the stalk to the posterior lobe. Interrupt the stalk and you cut both deliveries at once. [2]
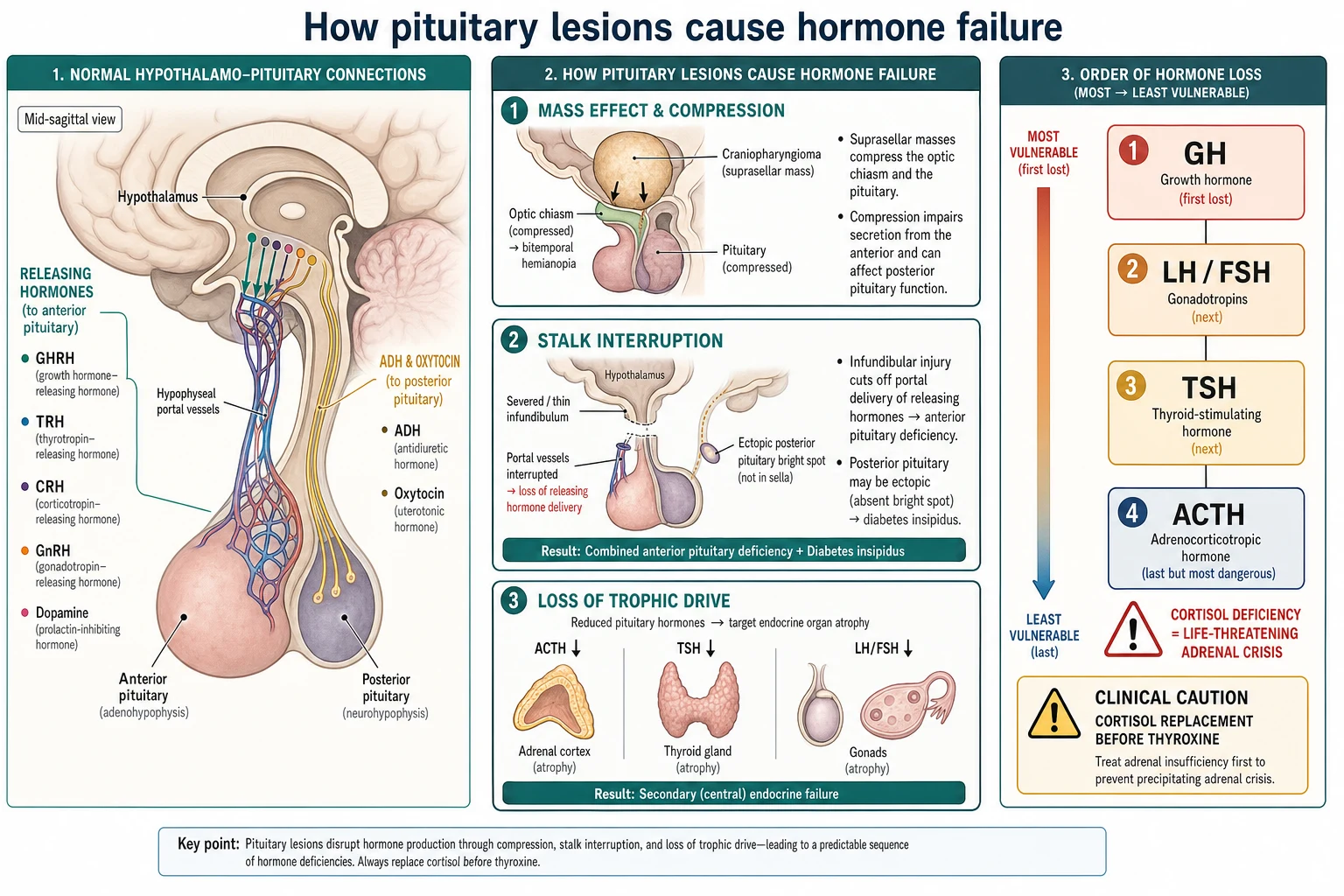
Lesions damage the axis in three ways. A mass such as a craniopharyngioma compresses the gland and the chiasm directly, causing hormone failure and bitemporal visual field loss together. A process that thins or severs the stalk — germinoma, Langerhans cell histiocytosis, or a congenital interruption — cuts the portal delivery of releasing hormones and blocks the transport of antidiuretic hormone, so combined anterior deficiency and central diabetes insipidus appear together. Loss of the trophic hormones then starves the target glands, which shrink. [5] [6]
The order in which anterior hormones fail reflects their vulnerability. Growth hormone and the gonadotrophins are the most fragile and are usually lost first; thyroid-stimulating hormone follows; adrenocorticotrophin, and therefore cortisol, is the most resilient and is usually last to go. This order matters because the least likely deficiency, cortisol deficiency, is also the most dangerous, and because starting thyroxine in a child whose adrenocorticotrophin reserve is marginal raises the metabolic rate and cortisol clearance and can unmask a crisis. [1] [10]
The posterior lobe adds its own logic. Antidiuretic hormone is made in the hypothalamus and stored in the posterior pituitary, seen on magnetic resonance imaging as the bright spot; when the stalk is interrupted the hormone cannot descend, the bright spot is lost or becomes ectopic, and the child develops central diabetes insipidus with dilute urine despite a rising plasma osmolality. [11] [2]
Clinical Presentation
A newborn with congenital hypopituitarism looks deceptively ordinary until the biochemistry is checked. The clues are hypoglycaemia — from combined growth hormone and cortisol deficiency — prolonged conjugated or unconjugated jaundice, a micropenis and undescended testes in a boy from gonadotrophin deficiency, and poor feeding or temperature instability. Any of these in combination should prompt an urgent paired glucose, cortisol and free thyroxine before treatment blurs the picture. [1]
Beyond the newborn period the presentation is dominated by growth. A child with growth hormone deficiency grows slowly, crossing height percentiles downwards with a low height velocity, and often looks younger and more chubby than peers with immature facial features. The other axes declare more quietly: fatigue and cold intolerance from central hypothyroidism, and absent or arrested puberty from gonadotrophin deficiency in an adolescent. [1] [8]
When a lesion is the cause, neurological features join the endocrine picture. A craniopharyngioma classically presents with the slow triad of growth failure, visual field loss and headache with raised intracranial pressure, sometimes with obesity and behavioural change from hypothalamic involvement. A germinoma often presents first with central diabetes insipidus — polyuria and polydipsia — with the anterior deficiencies and the visible mass following later. [5] [6]
Differential Diagnosis
The differential turns on separating a true pituitary deficiency from the far commoner benign mimics of slow growth and delayed puberty, and the biochemistry with imaging does the sorting. Constitutional delay of growth and puberty is the great mimic: the child is short and late but healthy, with a normal height velocity for bone age and a family history of late blooming, and the axes are intact. Familial short stature gives a short child growing steadily along a low percentile with normal timing. [8]
Points to hypopituitarism
- Height velocity falling, crossing percentiles downwards
- Neonatal hypoglycaemia, prolonged jaundice, micropenis
- Low free thyroxine with a non-raised thyroid-stimulating hormone
- Visual field defect, headache or diabetes insipidus
- Low IGF-1 with a subnormal growth hormone stimulation test
Points to a benign mimic
- Constitutional delay: healthy, late, normal velocity for bone age
- Familial short stature: steady low percentile, normal timing
- Primary hypothyroidism: raised thyroid-stimulating hormone
- Primary gonadal or adrenal failure: raised pituitary hormone
- Small-for-gestational-age without catch-up growth
The endocrine mimics are sorted by whether the pituitary hormone is high or low for its target. In central hypothyroidism the free thyroxine is low while the thyroid-stimulating hormone is low or inappropriately normal, whereas in primary hypothyroidism the thyroid-stimulating hormone is high. In central adrenal insufficiency the cortisol is low with a low or normal adrenocorticotrophin, whereas primary adrenal failure raises adrenocorticotrophin. The pituitary hormone that fails to rise when its target is low is the tell of a central problem. [1] [9]
Clinical & Bedside Assessment
The focused assessment starts with accurate growth data and a proper plot. Measure height and weight, calculate the height velocity over at least six months, plot the points on a growth chart with the mid-parental target range, and estimate the bone age from a hand and wrist radiograph, because a delayed bone age with a low velocity points away from constitutional delay towards a hormone deficiency. Record head circumference in an infant. [8]
Examine the eyes and the midline, because they carry the neurological clues. Test the visual fields to confrontation and examine the optic discs for the pallor and small size of optic nerve hypoplasia, which flags septo-optic dysplasia; look for a midline cleft lip or palate and a single central incisor that mark midline developmental defects. Assess the pubertal stage against the chronological age, and in a boy inspect the genitalia for a micropenis and undescended testes. [3] [2]
Read the fluid balance at the bedside when a lesion is suspected. A child with polyuria and polydipsia, a high plasma osmolality and dilute urine has central diabetes insipidus, and the danger is a child who also has cortisol deficiency, because cortisol is needed to excrete a water load and the diabetes insipidus may be masked until glucocorticoid replacement unmasks it. Chart the intake, output and daily weight. [11] [9]
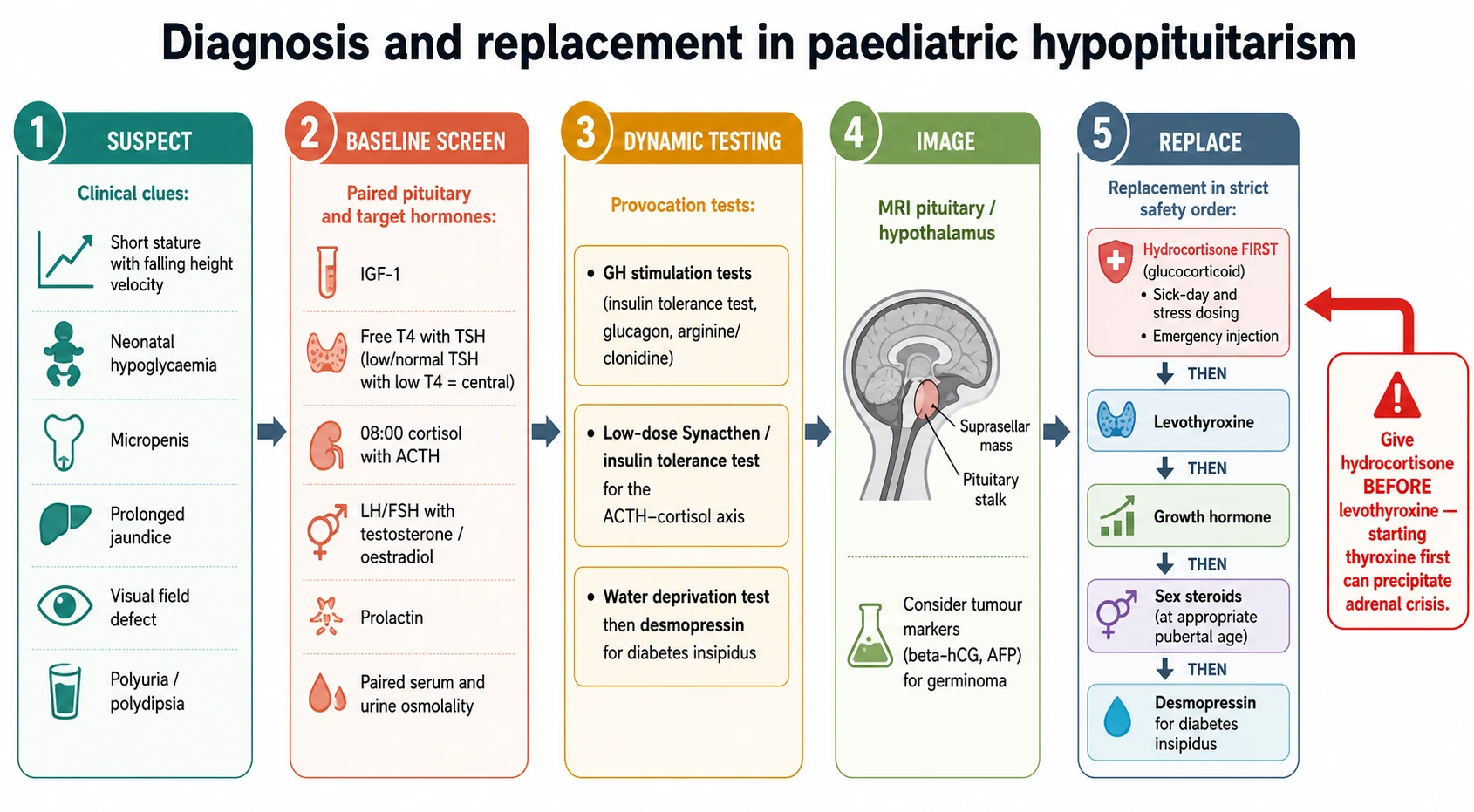
Investigations
The biochemical work-up pairs each pituitary hormone with its target and adds dynamic testing where a random level cannot decide. Draw an early-morning cortisol with adrenocorticotrophin, a free thyroxine with thyroid-stimulating hormone, IGF-1, the gonadotrophins with testosterone or oestradiol as age dictates, and prolactin, and pair a serum osmolality with a urine osmolality if diabetes insipidus is suspected. A low target hormone with a pituitary hormone that has not risen defines a central deficiency. [1] [8]
Growth hormone cannot be diagnosed on a random level because secretion is pulsatile, so a low IGF-1 with clinical growth failure is confirmed with growth hormone stimulation testing — an insulin tolerance test, or glucagon, arginine or clonidine stimulation — with a subnormal peak defining deficiency. The cortisol axis is confirmed with a low-dose Synacthen test or an insulin tolerance test when the morning cortisol is indeterminate. Central diabetes insipidus is confirmed with a formal water deprivation test followed by desmopressin, which concentrates the urine in central but not nephrogenic disease. [7] [11]
Imaging is the second half. A dedicated magnetic resonance imaging study of the hypothalamus and pituitary with contrast looks for a small or absent anterior gland, an interrupted stalk with an ectopic posterior bright spot, and any suprasellar mass; a thickened stalk with diabetes insipidus demands serum and cerebrospinal-fluid beta-human-chorionic-gonadotrophin and alpha-fetoprotein to catch a germinoma. In septo-optic dysplasia the imaging shows optic nerve hypoplasia and an absent septum pellucidum. [5] [6]
Management — Resuscitation
The resuscitation emergencies of hypopituitarism are hypoglycaemia and adrenal crisis, and both are treated before the diagnostic work-up is complete. A hypoglycaemic neonate or child with suspected hypopituitarism receives an intravenous glucose bolus and then stress-dose hydrocortisone, ideally after a critical sample for glucose, cortisol, growth hormone and insulin has been drawn at the point of hypoglycaemia. Waiting for confirmatory results before treating is unsafe. [1] [9]
An adrenal crisis in a child with central adrenal insufficiency presents with vomiting, lethargy, hypotension and hypoglycaemia, often triggered by an intercurrent illness, and the treatment is immediate parenteral hydrocortisone at a stress dose of about 50 to 100 mg per square metre as a bolus followed by a similar total across the day in divided doses, together with intravenous fluid and glucose. Unlike primary adrenal failure, central disease usually spares aldosterone, so the salt-wasting and hyperkalaemia of primary crisis are typically absent. [9] [10]
Every child on glucocorticoid replacement needs a sick-day plan and an emergency injectable. The family is taught to double or triple the oral hydrocortisone during febrile illness, to give intramuscular hydrocortisone and seek help if the child is vomiting or drowsy, and to carry written instructions and a medical alert. This education is part of the resuscitation strategy because most crises are prevented at home. [9] [10]
Management — Definitive & Stepwise
Definitive management replaces each deficient hormone and treats the underlying lesion, in a fixed safety order. Glucocorticoid comes first: hydrocortisone is given orally at a physiological dose of roughly 8 to 10 mg per square metre per day in three divided doses, titrated to clinical wellbeing and growth rather than to a blood level, with the sick-day and stress rules layered on top. Only once the cortisol axis is covered is levothyroxine started, dosed to bring the free thyroxine into the upper half of the reference range. [10] [9]
Growth hormone is then replaced by daily subcutaneous injection once deficiency is confirmed, at a starting dose of about 0.025 to 0.035 mg per kilogram per day, titrated to the height velocity and the IGF-1, and continued through childhood until final height is reached, after which adult reassessment decides on continuation. Cortisol cover must be secure before growth hormone begins, because growth hormone can lower cortisol availability by enhancing its metabolism. [7] [10]
Replacement in safe order
Step 1 — Glucocorticoid first
Hydrocortisone before anything else, with sick-day and stress dosing and an emergency injectable at home.
Step 2 — Levothyroxine
Started only after glucocorticoid is covered, titrated to the free thyroxine.
Step 3 — Growth hormone
Daily subcutaneous injection, titrated to the height velocity and IGF-1.
Step 4 — Sex steroids
Induce and progress puberty at an age matching peers to build growth and bone mass.
Step 5 — Desmopressin
For central diabetes insipidus, matched to thirst and paired osmolality.
Sex-steroid replacement is timed to physiology: puberty is induced with low-dose testosterone in a boy or oestradiol in a girl at an age matching peers, then titrated upwards over two to three years to complete pubertal development and support the pubertal growth spurt and bone mass. Central diabetes insipidus is treated with desmopressin, titrated carefully to thirst and to paired plasma and urine osmolality, with the constant caution that over-treatment causes hyponatraemia. The lesion itself is treated in parallel by the neurosurgical and oncology teams. [11] [6]
Specific Subtypes & Scenarios
Craniopharyngioma is the archetypal acquired cause. It is a benign but locally aggressive suprasellar tumour, classically cystic and calcified, that presents with the slow triad of growth failure, visual loss and raised intracranial pressure, and often with hypothalamic obesity. Treatment is surgical resection, sometimes with radiotherapy, and it usually leaves panhypopituitarism and diabetes insipidus that need lifelong replacement, with hypothalamic damage causing intractable obesity and behavioural change as the hardest long-term problem. [5]
Intracranial germinoma is the treatable malignancy that must not be missed. It arises in the midline, often in the suprasellar or pineal region, and characteristically causes central diabetes insipidus with anterior deficiencies and stalk thickening before a mass is obvious. It may secrete beta-human-chorionic-gonadotrophin, so serum and cerebrospinal-fluid markers are sent, and because germinoma is exquisitely radiosensitive and chemosensitive the prognosis is excellent when it is diagnosed early. [6]
Septo-optic dysplasia is the classic congenital midline disorder, defined by any two of optic nerve hypoplasia, midline forebrain abnormality such as an absent septum pellucidum, and pituitary hormone deficiency. It is often linked with HESX1 and related genes, and the endocrine phenotype ranges from isolated growth hormone deficiency to panhypopituitarism, so every child with optic nerve hypoplasia needs endocrine assessment and follow-up. [3] [2]
Pituitary stalk interruption syndrome ties the anatomy to the hormones. Magnetic resonance imaging shows a thin or absent stalk, a small anterior pituitary and an ectopic posterior bright spot, and the child has combined anterior deficiency, sometimes evolving over years, which is why an initially isolated growth hormone deficiency with this imaging picture warrants ongoing surveillance of the other axes. [4] [2]
Complications & Pitfalls
The complications of untreated hypopituitarism map to the missing hormones: hypoglycaemia and adrenal crisis from cortisol deficiency, growth failure and impaired final height from growth hormone deficiency, absent puberty and low bone mass from gonadotrophin deficiency, and the cognitive and metabolic effects of central hypothyroidism. The most dangerous is cortisol deficiency, because it is the least likely to be present yet the most rapidly lethal when it is. [1] [9]
The pitfalls are mostly errors of sequence and of screening. Starting thyroxine before glucocorticoid can precipitate an adrenal crisis and is the classic lethal error. Screening the thyroid with the thyroid-stimulating hormone alone misses central hypothyroidism, whose thyroid-stimulating hormone is not raised. Closing the file after one normal axis misses the evolving deficiencies of stalk interruption and post-irradiation disease, which appear over years. [10] [4]
Two further traps recur in the exam. Over-treating central diabetes insipidus with desmopressin causes dangerous hyponatraemia, so the dose is matched to thirst and osmolality. And forgetting that central adrenal insufficiency can mask diabetes insipidus means the polyuria appears only once hydrocortisone is started, catching the unwary team off guard. [11] [9]
Prognosis & Disposition
With prompt diagnosis and correct replacement the prognosis for most children with hypopituitarism is good: growth hormone restores growth towards the target height, glucocorticoid and thyroxine restore metabolic stability, and induced puberty and bone health follow. The limiting factors are the underlying cause and its own complications rather than the hormone replacement, which is well established and safe when supervised. [1] [7]
The prognosis of the lesions varies. Germinoma has an excellent outlook because it responds so well to radiotherapy and chemotherapy when caught early. Craniopharyngioma carries a good survival but a heavy morbidity burden from the panhypopituitarism, diabetes insipidus, visual loss and, above all, the hypothalamic obesity that resists treatment and dominates quality of life. Disposition is to a specialist paediatric endocrine service linked with neurosurgery, oncology, ophthalmology and genetics, with a structured transition to adult care. [5] [6]
Special Populations
Neonates and infants are the highest-stakes group because congenital hypopituitarism presents with hypoglycaemia that injures the developing brain and with a cortisol deficiency that can be fatal. The threshold for a critical sample and empirical stress hydrocortisone is low, and any infant with optic nerve hypoplasia, a midline defect or a micropenis with hypoglycaemia is assumed to have hypopituitarism until it is excluded. [1] [3]
Survivors of childhood cancer and cranial radiotherapy are a large and growing population at risk of evolving, dose-dependent hypopituitarism, with growth hormone the earliest and commonest loss. They need lifelong endocrine surveillance built into their survivorship care, because axes can fail years after treatment ends. [1] [5]
Adolescents in transition carry the twin burdens of pubertal induction and the handover to adult services, where growth hormone and other replacements are reassessed for the adult phase. Indigenous, migrant and remote-dwelling families may face barriers of distance and access to a specialist service, injectable growth hormone and reliable glucocorticoid supply, so a clear local emergency plan, telehealth support and a written sick-day protocol that travels with the child are essential. [7] [9]
Evidence, Guidelines & Regional Differences
The evidence base blends adult and paediatric guidance. The Endocrine Society guideline on hormonal replacement in hypopituitarism (Fleseriu and colleagues, 2016) sets the replacement principles, including the glucocorticoid-before-thyroxine rule, and its adrenal-insufficiency guideline (Bornstein and colleagues, 2016) governs the cortisol axis. The Pediatric Endocrine Society and Growth Hormone Research Society guidelines (Grimberg and colleagues, 2016; Collett-Solberg and colleagues, 2019) set the paediatric standards for diagnosing and treating growth hormone deficiency. [10] [7]
The paediatric-specific evidence draws on the developmental genetics of Kelberman and Dattani, the septo-optic dysplasia review of Webb and Dattani, and the work of Cerbone and Dattani on the evolution from isolated to combined deficiency, which underpins the surveillance principle. The lesions are covered by the craniopharyngioma primer of Müller and colleagues and the germinoma review of Liu and colleagues, and the water balance by the diabetes-insipidus review of Di Iorgi and colleagues. [2] [5]
Regional practice differs mainly in access and funding rather than in principle. Growth hormone is prescribed against country-specific criteria and funding rules, and in Australia and New Zealand suspected paediatric pituitary disease is referred to a tertiary paediatric endocrine service with linked neurosurgery, oncology and genetics. The diagnostic dynamic tests and the replacement order are internationally consistent, so the guidelines transfer well across settings. [7] [1]
Exam Pearls
Hold one sentence for the viva: a child with short stature and a falling height velocity, or a neonate with hypoglycaemia and a micropenis, has hypopituitarism until proven otherwise, and the rule that keeps them alive is to replace glucocorticoid before thyroxine. Say it early and the examiner knows you understand the danger. [1] [10]
State the frequently misremembered facts correctly. The anterior hormones usually fail in the order growth hormone, then gonadotrophins, then thyroid-stimulating hormone, then adrenocorticotrophin — so cortisol deficiency is last but most dangerous. Central hypothyroidism shows a low free thyroxine with a low or normal thyroid-stimulating hormone, so screen with the free thyroxine, not the thyroid-stimulating hormone alone. Growth hormone needs a stimulation test because secretion is pulsatile. [1] [7]
The high-yield lesion pairings: growth failure with visual field loss and a calcified cystic suprasellar mass is a craniopharyngioma; central diabetes insipidus with a thickened stalk and positive markers is a germinoma; optic nerve hypoplasia with an absent septum pellucidum is septo-optic dysplasia; a thin stalk with an ectopic posterior bright spot is pituitary stalk interruption syndrome. These pairings do most of the diagnostic work in the written and clinical exam. [5] [6]
Order of anterior pituitary hormone loss
Go Look For The Adenoma
Lost first and the most radiosensitive axis.
The gonadotrophins fail next, giving absent or arrested puberty.
Central hypothyroidism follows, with a low free thyroxine and a non-raised thyroid-stimulating hormone.
Last to fail but cortisol deficiency is the most dangerous, which is why glucocorticoid is replaced before thyroxine.
References
- [1]Higham CE; Johannsson G; Shalet SM Hypopituitarism. Lancet, 2016.PMID 27041067
- [2]Kelberman D; Dattani MT Hypothalamic and pituitary development: novel insights into the aetiology. Eur J Endocrinol, 2007.PMID 17785694
- [3]Webb EA; Dattani MT Septo-optic dysplasia. Eur J Hum Genet, 2010.PMID 19623216
- [4]Cerbone M; Dattani MT Progression from isolated growth hormone deficiency to combined pituitary hormone deficiency. Growth Horm IGF Res, 2017.PMID 29107171
- [5]Müller HL; Merchant TE; Warmuth-Metz M; et al Craniopharyngioma. Nat Rev Dis Primers, 2019.PMID 31699993
- [6]Liu AP; Takami H; Abdelbaki MS; et al Germinoma: Presentation, Management, and Recent Advances. Adv Cancer Res, 2025.PMID 41198339
- [7]Grimberg A; DiVall SA; Polychronakos C; et al Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency. Horm Res Paediatr, 2016.PMID 27884013
- [8]Collett-Solberg PF; Ambler G; Backeljauw PF; et al Diagnosis, Genetics, and Therapy of Short Stature in Children: A Growth Hormone Research Society International Perspective. Horm Res Paediatr, 2019.PMID 31514194
- [9]Bornstein SR; Allolio B; Arlt W; et al Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2016.PMID 26760044
- [10]Fleseriu M; Hashim IA; Karavitaki N; et al Hormonal Replacement in Hypopituitarism in Adults: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2016.PMID 27736313
- [11]Di Iorgi N; Napoli F; Allegri AE; et al Diabetes insipidus--diagnosis and management. Horm Res Paediatr, 2012.PMID 22433947