Paeds · endocrinology-diabetes-and-growth
Lipid disorders and familial hypercholesterolaemia
Also known as Familial hypercholesterolaemia · FH · Heterozygous familial hypercholesterolaemia · Homozygous familial hypercholesterolaemia · LDL receptor defect · Paediatric dyslipidaemia
Fellowship guide to lipid disorders and familial hypercholesterolaemia: the LDL receptor pathway and its genetic defects, the LDL-C thresholds that flag FH in a child, selective and universal screening, the secondary mimics, the statin-first drug ladder with ezetimibe and PCSK9 inhibitors, apheresis and newer agents for homozygous FH, and cascade screening of relatives.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single fact that organises everything is the raised LDL-C from birth. A child with familial hypercholesterolaemia is not sick in front of you; the artery is sick, slowly, over decades. That is why the disease is missed, why it is under-treated, and why the evidence for treating it in childhood is so important. Lowering LDL-C early can normalise the carotid artery wall, and twenty years of follow-up shows no growth or safety penalty. [2] [6]
This page covers the recognition and management of paediatric lipid disorders, with familial hypercholesterolaemia at the centre: the LDL receptor pathway and its genetic defects, the LDL-C thresholds that flag the disease, selective and universal screening, the secondary causes that mimic it, the statin-first drug ladder, apheresis and the newer agents for homozygous disease, cascade screening of relatives, and the transition of the adolescent into adult care. [1]
Overview & Definition
Paediatric lipid disorders are abnormalities of circulating lipids — cholesterol, triglycerides and their lipoprotein carriers — found in childhood. Most are common and mild, driven by diet, adiposity and inactivity, and they raise triglycerides and lower HDL more than they raise LDL. The lipid disorder that carries the heaviest lifetime risk and the strongest case for early drug treatment is familial hypercholesterolaemia, an inherited defect of the LDL receptor pathway that produces a high LDL-C from birth. [1]
Familial hypercholesterolaemia is an autosomal dominant condition in nearly all cases, though a rare autosomal recessive form exists (LDLRAP1). A pathogenic variant in one allele is enough to roughly double the LDL-C, so a parent and half of siblings and children share the diagnosis. The three genetic causes are loss-of-function variants in the LDL receptor gene (LDLR) in most cases, a defective apolipoprotein B that cannot bind the receptor (APOB), and a gain-of-function PCSK9 that destroys receptors faster than they are made. [2]
The clinically useful distinction at the bedside is between heterozygous and homozygous disease. A child with one pathogenic allele has heterozygous FH and a roughly one in 250 birth prevalence; untreated, they develop atherosclerotic cardiovascular disease in the fourth to sixth decade. A child with two pathogenic alleles has homozygous FH and a prevalence near one in 160,000 to 300,000; untreated, they develop tendon xanthomas, aortic stenosis and coronary disease in the first or second decade. The distinction matters because it changes the urgency and the drug ladder. [3]
Classification
The fastest way to classify a raised cholesterol in a child is to separate the primary (genetic) causes from the secondary (acquired) ones, because the work-up and the treatment diverge immediately. The figure below sets the genetic causes of familial hypercholesterolaemia against the acquired mimics, then gives the LDL-C thresholds that flag likely FH in a child. [2]
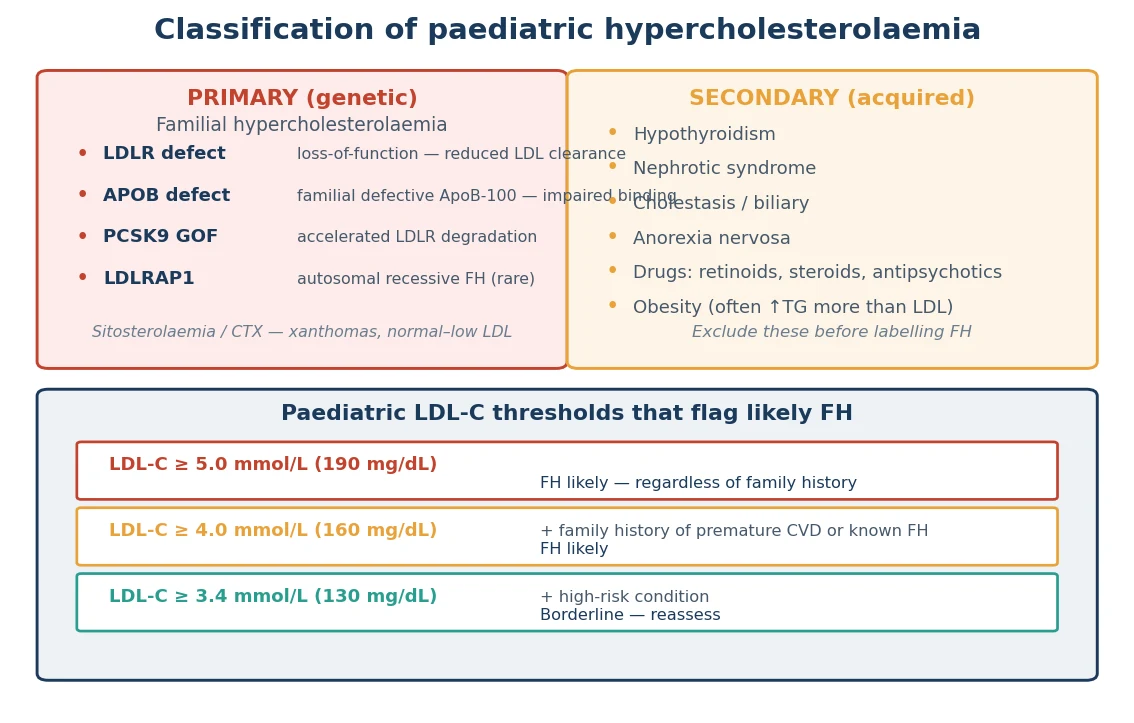
Primary (genetic) — FH
- LDLR loss-of-function (~60–80% of FH)
- APOB defect / familial defective ApoB-100 (~5–10%)
- PCSK9 gain-of-function (~1–2%)
- LDLRAP1 — autosomal recessive FH (rare)
- HeFH ~1 in 200–250; HoFH ~1 in 160,000–300,000
Secondary (acquired)
- Hypothyroidism — check TSH
- Nephrotic syndrome — proteinuria, low albumin
- Cholestasis / biliary obstruction
- Anorexia nervosa
- Drugs: retinoids, steroids, antipsychotics, thiazides
Two mimics deserve a name because they produce xanthomas with a near-normal LDL-C and would mislead a clinician who assumes FH. Sitosterolaemia is a defect of plant-sterol transport that deposits sterols in tendons and arteries; it is worsened, not helped, by standard low-cholesterol advice that increases plant-sterol intake, and it responds to ezetimibe. Cerebrotendinous xanthomatosis is a bile-acid synthesis defect that causes xanthomas with cataracts and neurological decline. Both are rare, but both sit on the differential of a xanthoma with a surprisingly modest LDL-C. [3]
Epidemiology & Risk Factors
Familial hypercholesterolaemia is the commonest inherited metabolic disorder that causes premature cardiovascular disease. The heterozygous form affects about one in 200 to 250 people in most populations, and the homozygous form about one in 160,000 to 300,000. The prevalence is higher in communities with a founder effect — French-Canadian, Afrikaner, Lebanese and Ashkenazi Jewish populations — and these pockets skew local screening and cascade programmes. [2]
The dominant inheritance is the key risk driver. An affected parent passes the variant to half their children, so a single index case should trigger cascade testing of the whole family. A sibling with FH, a parent with known FH, or a first-degree relative with premature cardiovascular disease each raise the chance that a given child carries the variant. Founder-effect ancestry adds a population-level risk. [1]
LIPID
The fingerprint of FH — an untreated LDL-C raised from childhood, dominant over triglycerides and HDL
Autosomal dominant; one affected parent means a one in two chance for each child and sibling
Men before 55 and women before 60 — draw a three-generation lipid family tree
One index case unlocks testing of the extended family — the cheapest way to find cases
Lifestyle, then a statin from age 8 to 10, then ezetimibe, then a PCSK9 inhibitor
The natural history without treatment is the whole reason the disease matters. Untreated heterozygous FH produces cardiovascular disease in the fourth to sixth decade; untreated homozygous FH produces childhood coronary and aortic-valve disease and is often fatal before the third decade. The risk is preventable with early, sustained treatment, which is why a raised LDL-C in an asymptomatic child is not a non-urgent incidental finding but a treatable exposure with a clock attached. [3] [6]
Pathophysiology
To understand why these children have a high LDL-C from birth, picture the hepatocyte as a recycling factory for cholesterol. An LDL particle in the bloodstream docks onto an LDL receptor on the liver cell surface; the receptor-ligand complex folds into the cell; the LDL is broken down in a lysosome; and the receptor is returned to the surface to catch another particle. That cycle clears most of the circulating LDL-C. [1]
A pathogenic variant breaks the cycle at one of three points. An LDLR loss-of-function variant removes or impairs the receptor, so LDL cannot be cleared. An APOB defect changes the binding site on the LDL particle itself, so the particle cannot dock onto an otherwise healthy receptor. A PCSK9 gain-of-function variant accelerates the receptor's destruction in the lysosome, so fewer receptors recycle to the surface. In each case the net effect is the same: circulating LDL-C rises. [2]
The raised LDL-C then does what cholesterol does when it sits in a vessel wall for decades. It is taken up by macrophages to form foam cells, the first step of an atheroma, and the deposit thickens the carotid intima and narrows the coronary lumen long before any symptom. In homozygous FH the LDL-C is so high and starts so early that it also deposits in tendons (tendon xanthomas), in the skin (cutaneous and tuberous xanthomas), in the cornea (arcus) and around the aortic valve, producing supravalvar aortic stenosis and coronary disease in childhood. [3]
The encouraging counterpoint is that the arterial clock can be slowed or reset if treatment starts early. In the PRECURSORY cohort, children who began a statin at a younger age achieved a normal carotid intima-media thickness, and twenty years of follow-up showed that sustained statin therapy preserved this benefit with no penalty to growth, puberty or the liver. Lowering LDL-C in childhood is not about a test result; it is about preventing the deposit before the vessel wall commits. [4] [6]
Clinical Presentation
Most children with familial hypercholesterolaemia are clinically silent. They are found by screening, by cascade testing of an affected family, or by a clinician who reaches for a lipid profile after taking a family history. The absence of symptoms is the rule, not the exception, and it is why the disease is underdiagnosed — the artery is sick while the child looks and feels well. [2]
The physical signs that support FH, when they appear, are cholesterol deposited in tissue. Tendon xanthomas — nodular thickening of the Achilles and the extensor tendons of the hand — are the classic sign and signal severe or homozygous disease. Xanthelasma (yellow cholesterol deposits in the eyelids) and juvenile corneal arcus are uncommon in heterozygous children and, when present, should prompt urgent specialist review. These signs are not normal in a child, and a candidate who dismisses them as cosmetic misses the diagnosis. [3]
The homozygous child presents differently. The LDL-C is markedly raised (above 10 mmol per litre), xanthomas appear in the first decade, and the disease declares itself through symptomatic coronary ischaemia, exertional syncope from supravalvar aortic stenosis, or a new murmur. Any child or adolescent with known or suspected FH who develops chest pain, exertional symptoms or a new murmur needs urgent cardiology assessment — this is the one part of the disease that is a genuine emergency. [3]
The family history is often the only clue that flags the asymptomatic heterozygous child. A first-degree relative with cardiovascular disease before 55 in men or 60 in women, a known FH diagnosis, tendon xanthomas, or a sudden cardiac death in the family should each trigger a lipid profile in the child. Drawing a three-generation lipid and cardiovascular family tree turns a chance finding into a systematic screen. [1] [5]
Differential Diagnosis
The differential of a raised LDL-C in a child splits into the secondary causes that raise cholesterol through another illness, and the rarer primary mimics that produce xanthomas without the typical LDL-C pattern. Reaching for the secondary causes first is the habit that stops a mislabelled diagnosis and a misdirected genetic test. [2]
Points to FH
- Untreated LDL-C ≥5.0 mmol/L, or ≥4.0 with family history
- Normal triglycerides and HDL
- Family history of premature CVD or known FH
- Tendon xanthomas (severe/HoFH)
- Positive genetic test (LDLR, APOB, PCSK9)
Points to a secondary cause
- Hypothyroidism — raised TSH, resolves with thyroxine
- Nephrotic syndrome — proteinuria, low albumin, oedema
- Cholestasis — raised ALP, GGT and bilirubin
- Drug exposure — retinoid, steroid, antipsychotic
- Raised LDL-C that falls when the cause is treated
The discriminating move is the secondary-cause screen, sent at the same time as the lipid profile: a thyroid-stimulating hormone, a renal panel with urinalysis and albumin, and liver function. If the LDL-C falls when hypothyroidism is treated, when the nephrotic syndrome remits, or when an offending drug is withdrawn, the diagnosis was secondary, not FH. The rarer mimics — sitosterolaemia and cerebrotendinous xanthomatosis — declare themselves through xanthomas with a modest LDL-C and need specific testing rather than a statin first. [3]
Polygenic hypercholesterolaemia is the other common alternative. It is milder, lacks the single-gene inheritance pattern, and does not carry the same lifetime risk or the same cascade-screening obligation. The distinction rests on the LDL-C level (polygenic is lower), the family history (polygenic is diluted), and the genetic test, which is negative for the three FH genes in polygenic disease. [1]
Clinical & Bedside Assessment
The assessment of a child with a raised LDL-C runs on two tracks: confirm the lipid abnormality and its likely cause, and assess the child and the family for the wider cardiovascular context. Begin with growth and pubertal stage, blood pressure, and body habitus, because the metabolic-syndrome overlap (obesity, raised blood pressure, a mixed lipid picture) both confounds the LDL-C and changes the lifestyle plan. [5]
Examine for the physical signs of severe FH — palpate the Achilles and extensor tendons for xanthomas, inspect the eyelids for xanthelasma and the cornea for arcus, and listen for an aortic murmur. These findings are uncommon in heterozygous children, but their presence upgrades the urgency. Plot the body mass index and the blood pressure on age-appropriate charts, because adiposity and hypertension compound the lifetime cardiovascular risk and are themselves treatable. [1]
In the adolescent, the assessment must include the psychosocial context: diet, physical activity, smoking and vaping, and alcohol. Lifestyle modification underpins every drug decision, and the same visit is the moment to begin contraception counselling for a girl who may be prescribed a teratogenic statin. A brief, non-judgemental adolescent psychosocial screen (the HEADSS framework) places the lipid result in the context of the young person's life rather than treating a number in isolation. [5]
Investigations
The core investigation is the lipid profile, and the question is whether the LDL-C crosses the FH threshold. A fasting or non-fasting panel gives the total cholesterol, LDL-C, HDL-C, non-HDL-C and triglycerides; lipoprotein(a) is added because a raised level adds independent cardiovascular risk and is found more often in FH families. The secondary-cause screen — thyroid-stimulating hormone, renal function and urinalysis, liver function — is sent at the same visit. [5] [9]
Investigation bundle for a child with a raised LDL-C
Lipid profile — total cholesterol, LDL-C, HDL-C, non-HDL-C, triglycerides
Lipoprotein(a) — adds independent risk, commoner in FH families
Secondary-cause screen — TSH, renal function, urinalysis and albumin, liver function
Apply the FH thresholds — LDL-C ≥5.0 mmol/L, or ≥4.0 with family history
Genetic testing — LDLR, APOB, PCSK9 (LDLRAP1 if homozygous or consanguinity)
Carotid intima-media thickness / echo — only in selected severe cases
The paediatric thresholds that flag likely FH are an untreated LDL-C at or above 5.0 mmol per litre (190 mg per decilitre), or at or above 4.0 mmol per litre (160 mg per decilitre) when there is a family history of premature cardiovascular disease or a known FH diagnosis in a parent. A borderline result is repeated, and a secondary cause is sought and treated before committing to a genetic test. A markedly raised LDL-C above 10 mmol per litre in a child places homozygous FH at the top of the differential. [2]
Genetic testing of LDLR, APOB and PCSK9 confirms the diagnosis, defines the variant, enables cascade testing of relatives, and supports pre-symptomatic testing of at-risk siblings. The LDLRAP1 gene is added when the pattern suggests autosomal recessive FH or there is parental consanguinity. Imaging — carotid intima-media thickness or echocardiography — is reserved for selected severe cases, such as homozygous FH, to gauge the burden of disease; it is not routine in the asymptomatic heterozygous child. [1]
Management — Resuscitation
Lipid disorders in children are rarely a resuscitation problem, and most of the work is deliberate and outpatient. The exception is the child or adolescent with homozygous FH who presents with acute chest pain, exertional syncope or a new murmur. That child may have premature coronary ischaemia or supravalvar aortic stenosis, and the situation is a cardiac emergency rather than a lipid problem. [3]
The immediate priorities in that scenario are paediatric cardiology and lipid-specialist involvement at once. Assess the child for coronary ischaemia with an electrocardiogram and troponin, arrange urgent echocardiography to look for aortic-valve and coronary involvement, and begin or escalate lipid-lowering therapy, including apheresis where it is available. The long-term plan follows once the child is stabilised, but the acute event is managed as any paediatric cardiac presentation. [3]
The more common situation — a markedly raised LDL-C found on a routine panel — is not an emergency. The exposure is chronic, measured in decades, and the right response is a careful, family-centred work-up and a treatment plan rather than an urgent dose. Confirm the result with a repeat and a secondary-cause screen, counsel the family, and start the definitive pathway in a planned way. [1]
The one acute medication risk to recognise is myopathy. A child already on a statin who presents with muscle pain or weakness needs a creatine kinase checked and the drug held until myopathy is excluded. Statin-related myopathy is rare in children, but the presentation of muscle symptoms on treatment is the one lipid-related problem that needs same-day assessment. [4] [6]
Management — Definitive & Stepwise
Once the diagnosis is confirmed, the definitive pathway aims at a single goal: lower the LDL-C early and keep it low, so the arterial deposit never gains its head start. The pathway builds from lifestyle to a statin, then to add-on drugs, then to the intensive therapies reserved for homozygous disease, with cascade screening of the family and structured transition running in parallel. [1]
Definitive pathway for confirmed paediatric FH
Lifestyle foundation — heart-healthy diet, activity, no smoking, weight management
Statin first-line — lowest licensed dose from age 8–10, titrated to target
Ezetimibe — add if LDL-C target not met on maximally tolerated statin
PCSK9 inhibitor — evolocumab / alirocumab for severe HeFH or HoFH
HoFH intensive therapy — LDL apheresis, lomitapide, evinacumab; consider liver transplant
Cascade screening — test all first-degree relatives
Annual review and structured transition to adult care
The lifestyle foundation is a heart-healthy diet low in saturated fat with adequate calories for growth, regular physical activity, avoidance of smoking and vaping, and weight management. Lifestyle underpins every drug decision, but it does not replace drug therapy in FH, because the genetic defect sets the LDL-C high from birth and diet alone cannot reach the target. [5]
A statin is the first-line drug and the evidence base in children is strong. Statins started at the lowest licensed dose from age 8 to 10 years and titrated to an LDL-C target normalise carotid intima-media thickness, and the twenty-year follow-up shows no penalty to growth, puberty or hepatic safety. Atorvastatin was shown to be effective and safe in a randomised trial of children with FH, and the class is now the standard of care for paediatric FH from the licensed age. [4] [12] [6]
Statin (e.g. atorvastatin, simvastatin, pravastatin, rosuvastatin)
The target for treated paediatric FH is an LDL-C ideally below 3.5 mmol per litre (about 135 mg per decilitre), and below 2.6 mmol per litre (100 mg per decilitre) in very-high-risk children. When the target is not met on a maximally tolerated statin, ezetimibe is added; ezetimibe monotherapy was shown to be effective and safe in children with heterozygous FH. For severe disease, a PCSK9 inhibitor (evolocumab or alirocumab) lowers LDL-C further and was effective in homozygous FH in the TESLA Part B trial. [11] [7] [1]
Specific Subtypes & Scenarios
The highest-yield scenarios for exams and for practice are the four below, because they are the ones that present, are missed, and are managed very differently. [1]
The heterozygous FH child in the school years is the commonest scenario. Detected by screening or cascade testing, the child is well, the LDL-C sits between 4 and 10 mmol per litre, and the triglycerides and HDL are normal. Management is lifestyle plus a statin from age 8 to 10, titrated to the target, with annual review. Treated this way, the child can expect a near-normal life expectancy. [2] [6]
The homozygous FH child or adolescent is the severe end of the spectrum. The untreated LDL-C is above 10 mmol per litre, xanthomas appear in the first decade, and coronary or aortic-valve disease follows. Management is intensive: LDL apheresis from early childhood, oral agents (ezetimibe plus a statin), and consideration of lomitapide, evinacumab (an ANGPTL3 inhibitor) or liver transplant. The newer agents have extended survival, but homozygous FH remains a severe, life-shortening disease. [3] [7]
The child with a secondary cause masquerading as FH is the diagnostic pitfall. Hypothyroidism, nephrotic syndrome, cholestasis or a drug (a retinoid, a steroid, an antipsychotic) raises the LDL-C, and the abnormality resolves when the underlying disorder is treated. The FH genetic test is negative, and the family is spared a mislabelled inherited diagnosis and a needless cascade screen. [5]
The adolescent girl with FH on a statin is the transition scenario that examiners probe. Statins are teratogenic, so reliable contraception is counselled before the drug is started, and the statin is stopped 4 to 6 weeks before a planned pregnancy. Lipid lowering is generally deferred during pregnancy and breastfeeding, because the maternal cardiovascular risk of a short treatment gap is outweighed by the fetal risk of the drug. [1] [9]
Complications & Pitfalls
The complications divide into those of the untreated disease and those of its treatment. Untreated FH drives premature atherosclerotic cardiovascular disease — myocardial infarction and stroke — and, in homozygous FH, supravalvar aortic stenosis and aortic-valve disease that declare in childhood. The psychological burden of a lifelong inherited diagnosis, and of the family screening it triggers, is a real complication and a reason for a thoughtful, non-stigmatising conversation. [2] [3]
The treatment complications are mostly mild and rare in children. Statin-related transaminitis and myopathy occur, and the relevant clinical move is to check creatine kinase and liver enzymes when a child on a statin reports muscle symptoms or fatigue, and to hold the drug until myopathy is excluded. PCSK9 inhibitors cause injection-site and flu-like reactions, and ezetimibe is generally well tolerated. The teratogenicity of statins in pregnancy is the treatment complication with the highest stakes, and it is managed by contraception counselling and pre-conception planning. [6] [7]
Disease pitfalls
- Missing FH because the child looks and feels well
- Assuming childhood is safe and deferring the statin to adulthood
- Labelling FH before excluding a secondary cause
- Overlooking homozygous FH behind 'incidental' xanthomas
Treatment pitfalls
- Prescribing a statin without contraception counselling in an adolescent girl
- Chasing the LDL-C number and ignoring growth and adherence
- Forgetting cascade screening of the extended family
- Withholding an effective drug for fear of harms the evidence does not show
The most consequential diagnostic pitfall is to attribute a high LDL-C to FH before excluding the secondary causes. Treating a raised LDL-C with a statin while leaving an unrecognised hypothyroidism or nephrotic syndrome untreated is a double error: the statin is unnecessary and the real disease is missed. The countermeasure is the secondary-cause screen at the first visit, interpreted alongside the lipid profile. [5]
The most consequential management pitfall is the mirror image: withholding an effective statin from a child with FH because of an unfounded fear of harm. The evidence is clear that statins started at the licensed age and dose are safe over two decades and reset the vascular clock, so the child who is denied treatment pays the price in arterial deposits that cannot later be undone. The habit that serves the child is to treat to target, monitor growth and adherence, and reassess at every visit. [4] [6]
Prognosis & Disposition
With early diagnosis and disciplined treatment, a child with heterozygous FH can expect a near-normal life expectancy. The single biggest determinant of outcome is whether treatment starts in childhood rather than after a first cardiovascular event, because the atherosclerotic deposit accumulates silently for decades before symptoms and can be prevented but not easily reversed. The PRECURSORY and twenty-year follow-up data underpin the confidence to treat early. [4] [6]
Homozygous FH remains a severe disease. Even with apheresis and the newer agents (lomitapide and evinacumab), childhood cardiovascular mortality is a real risk and survival is not normalised. The prognosis for these children depends on early, intensive, multidisciplinary care and on access to apheresis, and it is the area where the gap between best and worst care is widest. [3] [7]
The long-term safety of childhood statin therapy is now established over twenty years of follow-up, with no signal for impaired growth, delayed puberty or hepatic harm. This evidence is what allows the clinician to recommend a statin to a well child and a worried family with confidence, and to keep the child on treatment through adolescence and into adulthood. [6]
Disposition is lifelong. The child is reviewed annually by a lipid specialist, the lipid panel is checked every 6 to 12 months, the growth and pubertal stage are plotted, and the LDL-C target is reassessed. Cascade screening of the extended family is completed, and a structured transition to adult care in late adolescence carries the dosing, the monitoring and the counselling forward. The plan that travels with the patient is the measure of good FH care. [1]
Special Populations
The affected family is the unit of care, not just the index child. Cascade testing of first-degree relatives is the cornerstone, because each first-degree relative has a one in two chance of carrying the variant. The child of an affected parent is tested from around age 2 years, and siblings are offered pre-symptomatic genetic testing once the family variant is known. One index case, worked up properly, can identify and treat a whole family. [2]
The adolescent girl at transition needs contraception counselling before a statin is prescribed, a plan to stop the statin 4 to 6 weeks before a planned pregnancy, and a clear understanding that lipid lowering is deferred during pregnancy and breastfeeding. The same visit is the moment to address adherence, the transition to adult care, and the young woman's understanding of her own cardiovascular risk. [1] [9]
Australia and Aotearoa New Zealand manage familial hypercholesterolaemia through specialist lipid clinics and the national FH initiatives, with cascade screening coordinated across families and primary care. The Australasian guidance aligns with the ESC/EAS framework: lifestyle, a statin from age 8 to 10, add-on ezetimibe and PCSK9 inhibitors, and apheresis for homozygous FH through the tertiary paediatric centres. Access to PCSK9 inhibitors and apheresis is the main equity challenge in remote and Indigenous communities. [1] [8]
Indigenous, migrant and remote populations face both a higher founder-effect prevalence in some communities and the logistics of accessing specialist services and apheresis. Universal lipid screening and cascade testing are the great equalisers here, but they only work when the result is acted on, which depends on follow-up, transport and culturally safe counselling. Equity of access to a lipid specialist and to the drug ladder is a determinant of long-term outcome. [2]
Evidence, Guidelines & Regional Differences
The guideline base for paediatric familial hypercholesterolaemia rests on the European Society of Cardiology and European Atherosclerosis Society dyslipidaemia guidelines — the 2016 edition and its 2019 update — and on the EAS consensus on FH in children and adolescents. The 2018 AHA/ACC cholesterol guideline sets the North American standard. Together they converge on the LDL-C thresholds, the statin-first drug ladder, and the principle of treating the asymptomatic child. [1] [8] [9] [10]
20-Year Follow-up of Statins in Children with FH (Luirink 2019)
N Engl J Med
Long-term follow-up of a cohort of children with familial hypercholesterolaemia treated with a statin from childhood, comparing carotid intima-media thickness and safety over twenty years with unaffected siblings.
Key finding
Sustained statin therapy from childhood preserved a near-normal carotid intima-media thickness into adulthood, with no signal for impaired growth, delayed puberty or hepatic harm.
Practice change
The long-term safety evidence supports starting a statin at the licensed age in children with FH and sustaining it through adolescence and transition.
The screening debate sits between a selective strategy and a universal one. The American Academy of Pediatrics position argues for selective screening of children aged 2 to 10 with a family history or other risk factors, while the NHLBI integrated guideline recommends a universal lipid screen at age 9 to 11 years and again at 17 to 21. The two strategies agree that a child with a family history of premature cardiovascular disease must be tested, and they differ on whether to test everyone. [5] [10]
The statin evidence in children transformed the field. The atorvastatin trial showed efficacy and safety in children with FH, the PRECURSORY cohort showed that earlier statin normalised carotid intima-media thickness, the twenty-year follow-up confirmed durable benefit with no safety penalty, and the ezetimibe monotherapy trial established the add-on option. The PCSK9 inhibitors extended the ladder for severe and homozygous disease. [12] [4] [6] [11] [7]
The remaining controversies are practical: the optimal paediatric LDL-C target and how tightly to chase it, the exact age and dose to start a statin, the place of PCSK9 inhibitors and newer agents in children, and the relative value of universal versus cascade screening. A fellowship candidate names the debate rather than pretending it is settled, and anchors every recommendation in the evidence and guideline base. [1] [9]
Exam Pearls
One-sentence answer: the approach to a child with a raised LDL-C
A child with an untreated LDL-C at or above 5.0 mmol per litre, or at or above 4.0 mmol per litre with a family history, has familial hypercholesterolaemia until proven otherwise: confirm with a repeat lipid profile and a secondary-cause screen, test for LDLR, APOB and PCSK9, exclude hypothyroidism, nephrotic syndrome, cholestasis and a drug cause, then begin lifestyle and a statin from age 8 to 10, titrated to an LDL-C target ideally below 3.1 mmol per litre, with cascade screening of the family and contraception counselling for the adolescent girl.
Epidemiology and thresholds
- HeFH ~1 in 200–250; HoFH ~1 in 160,000–300,000
- FH-likely: LDL-C ≥5.0 mmol/L (190), or ≥4.0 (160) with family history
- LDLR (~60–80%), APOB (~5–10%), PCSK9 GOF (~1–2%)
- HoFH: LDL-C >10 mmol/L; childhood xanthomas and CVD
The drug ladder
- Statin first-line from age 8–10, lowest licensed dose, titrate to target
- Ezetimibe if target not met (Kusters 2015)
- PCSK9 inhibitor (evolocumab) for severe / HoFH (Raal 2015)
- HoFH: apheresis, lomitapide, evinacumab; consider liver transplant
Targets, screening and safety
- LDL-C target ideally <3.5 mmol/L (135 mg/dL); <2.6 (100) if very-high-risk
- Cascade-test every first-degree relative (1 in 2 chance)
- 20-year statin follow-up: no growth, puberty or hepatic harm (Luirink 2019)
- Statin is teratogenic — contraception for adolescent girls
Frequently misremembered facts, stated correctly: a raised LDL-C in a child is a chronic exposure, not an emergency. Childhood tendon xanthomas and juvenile corneal arcus are not normal and signal severe or homozygous FH. Statins are not contraindicated in children when started at the licensed age and dose — the twenty-year evidence is reassuring. And the disease is inherited in an autosomal dominant pattern, so one index case should trigger cascade testing of the whole family rather than treatment of the child alone. [6]
The lesion-to-sign pairings are the fastest route to a bedside answer and the highest-yield material for a written or oral question: a raised LDL-C with a normal triglyceride points to FH; childhood tendon or cutaneous xanthomas point to homozygous FH or sitosterolaemia; a raised LDL-C that resolves when thyroxine is started or a steroid is tapered points to a secondary cause, not FH; and a positive family history of premature cardiovascular disease in a parent points to heterozygous FH in the child. [3] [5]
References
- [1]Wiegman A; Gidding SS; Watts GF; et al Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J, 2015.PMID 26009596
- [2]Nordestgaard BG; Chapman MJ; Humphries SE; et al Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J, 2013.PMID 23956253
- [3]Cuchel M; Bruckert E; Ginsberg HN; et al Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. Eur Heart J, 2014.PMID 25053660
- [4]Rodenburg J; Vissers MN; Wiegman A; et al Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation, 2007.PMID 17664376
- [5]Daniels SR; Greer FR; Committee on Nutrition Lipid screening and cardiovascular health in childhood. Pediatrics, 2008.PMID 18596007
- [6]Luirink IK; Wiegman A; Kusters DM; et al 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N Engl J Med, 2019.PMID 31618540
- [7]Raal FJ; Honarpour S; Wasserman SM; et al Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet, 2015.PMID 25282520
- [8]Catapano AL; Graham I; De Backer G; et al 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur Heart J, 2016.PMID 27567407
- [9]Mach F; Baigent C; Catapano AL; et al 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J, 2020.PMID 31504418
- [10]Khera A; Heidenreich PA The New 2018 Cholesterol Guidelines. Circulation, 2019.PMID 30586686
- [11]Kusters DM; Oosterloo J; Wiegman A; et al Efficacy and safety of ezetimibe monotherapy in children with heterozygous familial or nonfamilial hypercholesterolemia. J Pediatr, 2015.PMID 25841542
- [12]McCrindle BW; Ose L; Marais AD Efficacy and safety of atorvastatin in children and adolescents with familial hypercholesterolemia or severe hyperlipidemia: a multicenter, randomized, placebo-controlled trial. J Pediatr, 2003.PMID 12915827