Paeds · endocrinology-diabetes-and-growth
Monogenic diabetes and neonatal diabetes
Also known as Monogenic diabetes · Maturity-onset diabetes of the young · MODY · Neonatal diabetes mellitus · NDM · Transient neonatal diabetes · Permanent neonatal diabetes · KATP channel neonatal diabetes · GCK-MODY · HNF1A-MODY
Fellowship guide to monogenic diabetes and neonatal diabetes: the single-gene beta-cell defects that masquerade as type 1 or type 2 diabetes, the neonatal diabetes that appears under six months and is almost never autoimmune, the GCK and HNF1A/HNF4A MODY subtypes that change the drug, the KATP channel mutations that switch a child from insulin to an oral sulfonylurea, and the genetic pathway from suspicion to a genotype-matched treatment.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The idea that organises the whole topic is a single-gene beta-cell defect that masquerades as type 1 or type 2 diabetes. Because the problem sits in one gene, the family history runs in an autosomal-dominant line, the islet autoantibodies are absent, the C-peptide survives beyond any honeymoon, and — critically — the correct treatment is set by which gene is at fault. Glucokinase-MODY rarely needs a drug at all, the HNF1A and HNF4A forms are exquisitely sensitive to a low-dose sulfonylurea, and the potassium-channel neonatal diabetes responds better to oral glibenclamide than to insulin. [1] [4]
This page covers the suspicion, confirmation and gene-matched treatment of monogenic diabetes in children: the neonatal diabetes that appears under six months, the GCK and HNF1A/HNF4A MODY subtypes that change the drug, the HNF1B form that brings the kidneys and pancreas with it, the genetic testing pathway, and the lifelong team care that follows a correct molecular label. It links to the dedicated leaves for type 1 and type 2 diabetes rather than repeating their full pathways. [2]
Overview & Definition
Monogenic diabetes is diabetes arising from a mutation in a single gene that governs beta-cell development, insulin production, or the glucose-sensing and secretion machinery. It accounts for an estimated one to four percent of childhood diabetes, although most affected children are mislabelled as having type 1 or type 2 for years before the molecular diagnosis is made. [1] [5]
The clinically useful distinction at presentation is between the two broad monogenic families. Neonatal diabetes mellitus is hyperglycaemia requiring insulin that begins in the first six months of life, a window in which autoimmune type 1 diabetes is vanishingly rare, so any diabetes here is presumed monogenic until genetics proves otherwise. Maturity-onset diabetes of the young is the monogenic diabetes of later infancy, childhood and adolescence, classically autosomal dominant, antibody-negative, and associated with preserved endogenous insulin. [2] [7]
Why the distinction from type 1 and type 2 matters is purely therapeutic. Type 1 diabetes demands insulin from the outset. Type 2 diabetes is managed with lifestyle and metformin, then escalating therapy. Monogenic diabetes is managed by gene: GCK-MODY usually needs no drug, HNF1A and HNF4A MODY respond to a sulfonylurea, and potassium-channel neonatal diabetes shifts from insulin to glibenclamide. A wrong label means a wrong drug, decades of unnecessary injections, and avoidable hypoglycaemia. [1] [4]
Classification
Classify monogenic diabetes first by the age window, then by the gene. The figure splits neonatal diabetes into its transient and permanent forms, and maturity-onset diabetes of the young into its common subtypes, then sets each against type 1 and type 2 diabetes. [1]
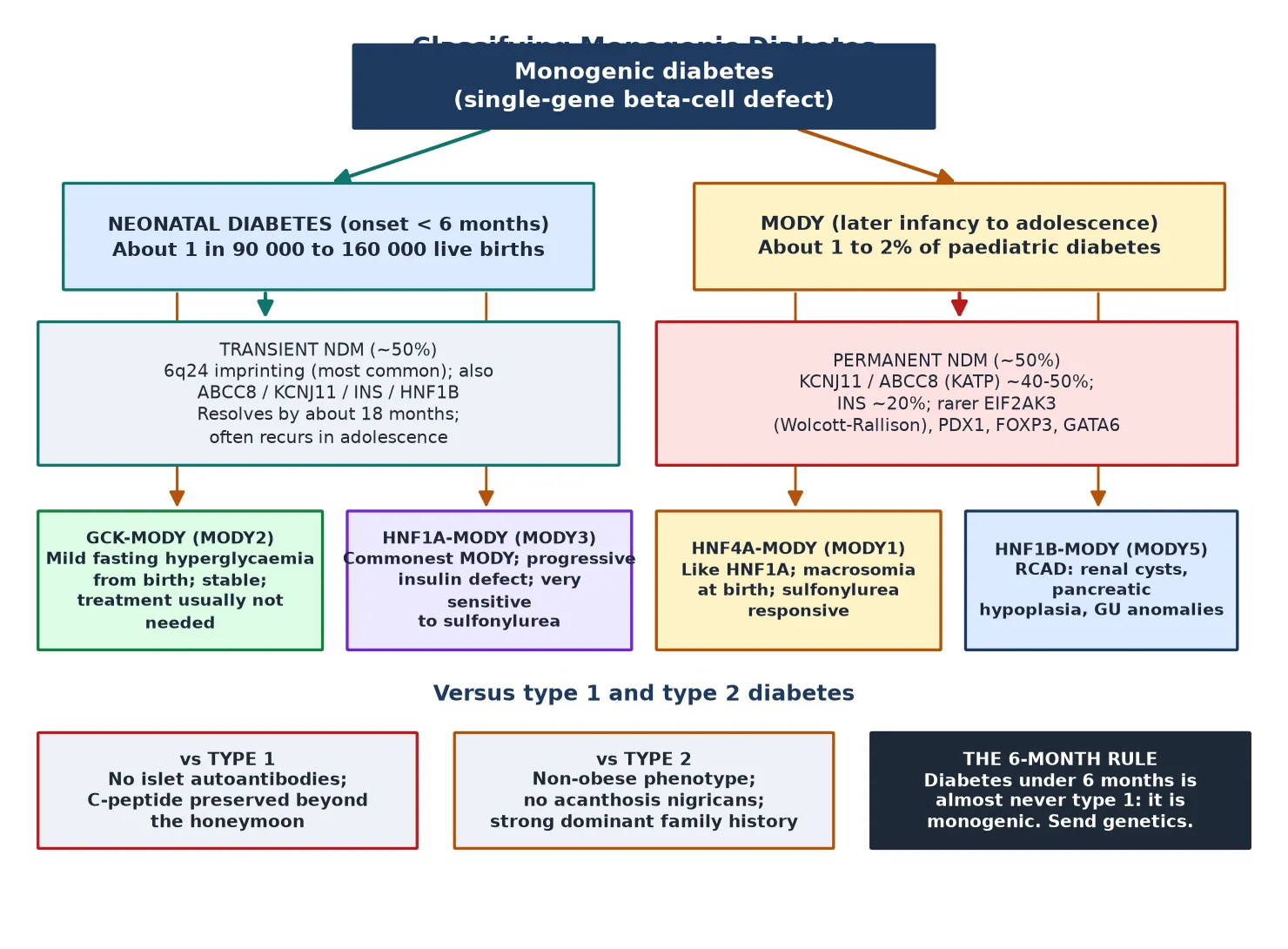
Neonatal diabetes (<6 months)
- Onset in the first six months of life
- Almost never autoimmune type 1
- Split into transient (often 6q24) and permanent (often KCNJ11/ABCC8)
- Genetic testing is mandatory and changes treatment
MODY (later infancy to adolescence)
- Autosomal-dominant family history across generations
- Antibody-negative with preserved C-peptide
- GCK is mild and stable; HNF1A/HNF4A are progressive
- HNF1B brings renal cysts and pancreatic hypoplasia
Neonatal diabetes mellitus itself splits into two natural histories that share a presentation. Transient neonatal diabetes remits, usually by around eighteen months, but frequently recurs in adolescence or adulthood, so the word transient describes the course, not the risk. Permanent neonatal diabetes never remits. Roughly half of permanent cases trace to activating mutations in the ATP-sensitive potassium-channel genes KCNJ11 and ABCC8, with INS mutations the next most common, and rarer syndromic genes such as EIF2AK3 (Wolcott-Rallison), PDX1, FOXP3 and GATA6 accounting for the rest. [7] [10]
The MODY subtypes each carry a recognisable signature. GCK-MODY, the glucokinase form, is a mildly raised, stable fasting glucose present from birth that seldom causes complications and seldom needs treatment. HNF1A-MODY and the rarer HNF4A-MODY are progressive insulin-secretory defects that are strikingly sensitive to low-dose sulfonylureas and carry a real microvascular risk over time. HNF1B-MODY is the multi-system form, pairing diabetes with renal cysts, pancreatic hypoplasia and genitourinary anomalies — the so-called renal cysts and diabetes syndrome. [1] [9]
Epidemiology & Risk Factors
Monogenic diabetes is uncommon but not rare: population estimates place it at one to four percent of all childhood diabetes, and registry work from the SEARCH study found that the great majority of children with HNF1A, HNF4A or glucokinase mutations had been clinically labelled as type 1 or type 2 before molecular testing. The under-diagnosis is the central epidemiological fact, because every missed case carries a wrong drug. [5]
Neonatal diabetes is rarer still, with an estimated incidence around one in ninety thousand to one in one hundred and sixty thousand live births, split roughly equally between transient and permanent forms. A recent Italian national survey tracking two decades of cases showed that as genetic testing widened, the recognised genetic and phenotypic spectrum expanded, with an increasing share of permanent cases attributable to potassium-channel mutations amenable to sulfonylurea treatment. [11]
The strongest risk factor is a positive family history in a dominant pattern. Because most MODY is autosomal dominant, a parent and child both labelled diabetic — especially both on insulin with negative antibodies — should prompt testing. The history must be taken across three generations, because the mild GCK form is easily missed in older relatives who never knew they had a raised glucose. Consanguinity raises the chance of the rarer recessive neonatal diabetes genes such as EIF2AK3 and PDX1. [1] [12]
Mnemonic devices help carry the suspicion at the bedside. The combination of a young age at onset, a lean phenotype, an antibody-negative screen, a measurable C-peptide beyond the honeymoon, and a dominant family history should trigger a referral for genetic testing in any child or adolescent with diabetes. [2] [6]
MONOGENIC
Maturity-onset diabetes of the young and neonatal diabetes mellitus are the two faces
Diabetes in the first six months is monogenic until genetics says otherwise
Islet autoantibodies are absent, separating it from autoimmune type 1
An autosomal-dominant line across three generations is the classic clue
A targeted monogenic panel or whole-exome sequencing confirms the gene
C-peptide is preserved beyond the honeymoon, unlike type 1
A lean phenotype with no acanthosis nigricans argues against type 2
The genotype sets the drug: no drug, sulfonylurea, or glibenclamide
Test first-degree relatives and offer genetic counselling
Pathophysiology
To see why a single gene causes diabetes, picture the beta cell as a glucose-driven machine: glucose enters, raises the ratio of ATP to ADP inside the cell, the ATP-sensitive potassium channel closes, the membrane depolarises, calcium enters, and insulin is released. Each monogenic gene breaks one step in that chain, which is why the disease looks different for each gene and why the treatment is gene-specific. [3] [10]
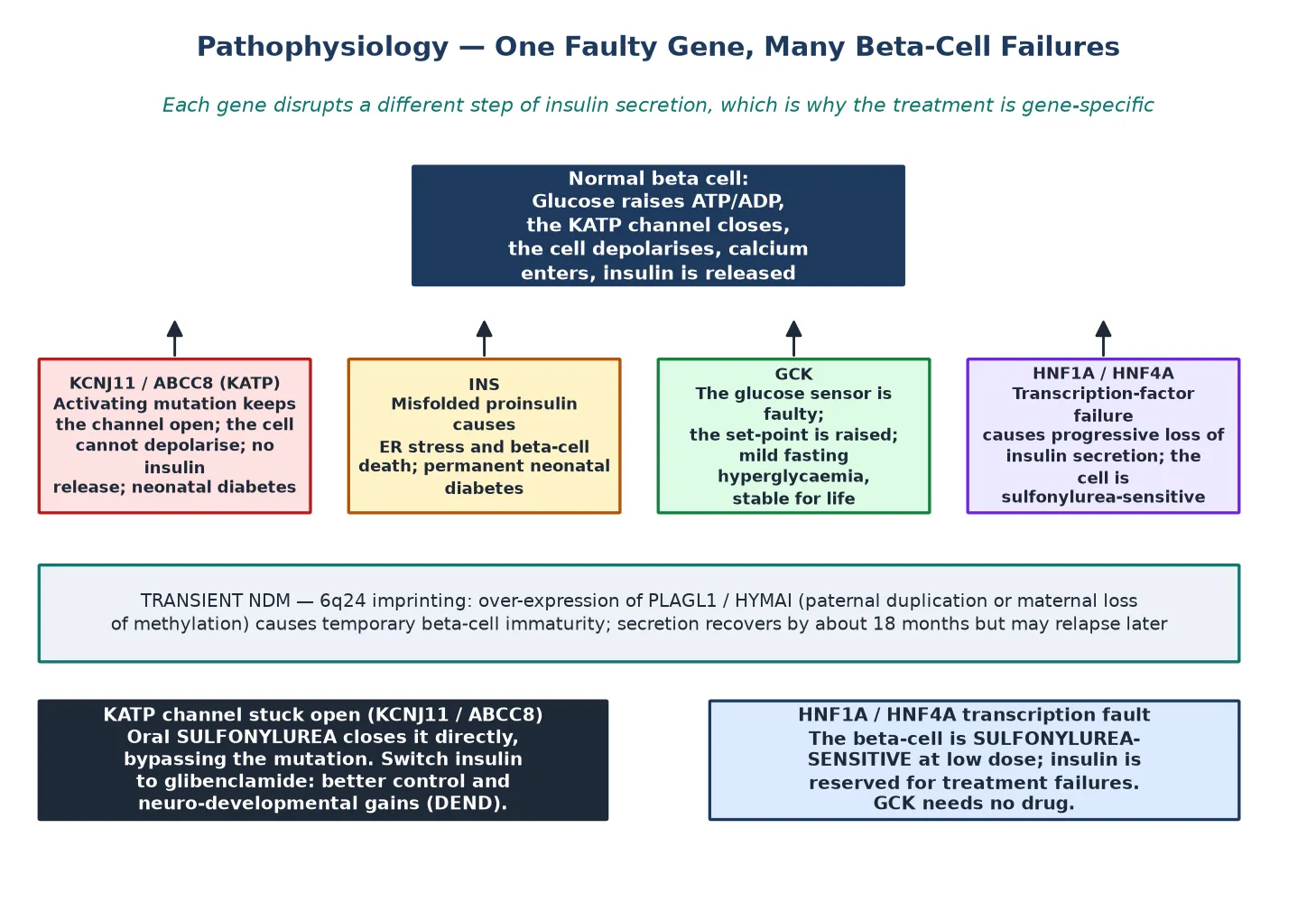
The potassium-channel genes are the archetype. An activating mutation in KCNJ11, which encodes the Kir6.2 subunit, or in ABCC8, which encodes the sulfonylurea receptor subunit, holds the channel permanently open. The beta cell cannot depolarise, calcium cannot enter, and insulin is not released — producing permanent neonatal diabetes. The same mutations can also affect neuronal potassium channels, which explains the developmental delay, epilepsy and muscle weakness of the DEND syndrome seen in some affected children. [3] [10]
The therapeutic corollary is the whole clinical point. A sulfonylurea such as glibenclamide binds the sulfonylurea receptor and closes the channel directly, bypassing the faulty ATP-sensing step and restoring insulin release. That is why a child with a potassium-channel mutation can be switched from insulin to an oral drug — and why the switch usually improves not only glycaemia but also the neurological features, because the same channel is at fault in the brain. The landmark trial of switching from insulin to oral sulfonylureas in Kir6.2 neonatal diabetes established this as a standard of care. [4]
INS mutations work through a different mechanism. Misfolded mutant proinsulin accumulates in the endoplasmic reticulum, triggering ER stress and progressive beta-cell death, which is why INS causes permanent neonatal diabetes with no sulfonylurea role — the cells are gone, not just silent. GCK mutations raise the glucose set-point: the beta cell still works, but only at a higher glucose, producing the mild, stable, fasting hyperglycaemia that defines glucokinase-MODY from birth and rarely progresses. [1] [8]
Channel open (KCNJ11/ABCC8)
- Activating mutation keeps the KATP channel open
- Beta cell cannot depolarise or release insulin
- Oral glibenclamide closes the channel directly
- Switch from insulin; neurological gain in DEND
Beta-cell death (INS)
- Misfolded proinsulin causes ER stress
- Progressive beta-cell loss
- Permanent neonatal diabetes
- Insulin required; no sulfonylurea role
Raised set-point (GCK)
- Faulty glucose sensor raises the threshold
- Mild, stable fasting hyperglycaemia from birth
- Seldom progresses or causes complications
- Usually needs no drug at all
Transcription failure (HNF1A/HNF4A)
- Progressive loss of insulin secretion
- Exquisitely sulfonylurea-sensitive
- Real microvascular risk over time
- Insulin reserved for treatment failures
The transient neonatal diabetes mechanism is separate again. Most transient cases arise from over-expression of imprinted genes at the 6q24 locus — either through paternal duplication or through loss of maternal methylation — which over-expresses PLAGL1 and HYMAI and produces a temporary beta-cell immaturity. Insulin secretion recovers by around eighteen months as the beta cell matures, but the predisposition to diabetes recurs in adolescence or adulthood, which is why transient is a description of the course rather than a reassurance. [10] [11]
Clinical Presentation
A neonate with diabetes typically presents in the first weeks to months of life with hyperglycaemia found during investigation of poor growth, dehydration, or sepsis-like illness, often in a neonatal or intensive care setting. The glucose is high, insulin is required, and the baby is small for gestational age in many cases because foetal insulin deficiency limits growth. Ketosis may be present, but the full ketoacidotic picture of autoimmune type 1 is less common in this age group. [7] [11]
The syndromic neonatal diabetes genes declare themselves through their extra-pancreatic features. A child with Wolcott-Rallison syndrome from biallelic EIF2AK3 mutations has neonatal or early-infantile diabetes together with epiphyseal dysplasia, recurrent hepatic dysfunction and failure to thrive. PDX1 mutations may pair neonatal diabetes with varying exocrine pancreatic insufficiency. The potassium-channel genes bring the neurological features of DEND — developmental delay, epilepsy and muscle weakness — which often dominate the early clinical picture before the diabetes is recognised as monogenic. [12] [10]
An older child or adolescent with MODY presents differently and is usually mislabelled first. The glucokinase form is often found incidentally on a screening glucose, runs a stable, mild course, and never causes the osmotic symptoms or ketoacidosis of type 1. The HNF1A and HNF4A forms present with progressive hyperglycaemia, may be detected through glycosuria at a school medical or during intercurrent illness, and are frequently mistaken for type 1 because of the age — or for type 2 because of the absence of autoimmunity. HNF1B-MODY may present through the renal tract, with hypoplastic or cystic kidneys, or through recurrent pancreatitis from pancreatic hypoplasia. [1] [5]
The history and phenotype that should trigger suspicion are the discriminators against type 1 and type 2. A dominant family history across three generations, an antibody-negative screen, a C-peptide that persists well beyond any honeymoon, a lean body habitus without acanthosis nigricans, and a glucose that is unexpectedly stable or mild all point toward monogenic disease. The combination, even without a named gene, is the trigger for referral. [2] [6]
Differential Diagnosis
The differential is between monogenic diabetes and the common forms it masquerades as. Type 1 diabetes is autoimmune, antibody-positive, and loses C-peptide over time, so a child with persistent measurable C-peptide years after diagnosis and negative antibodies is not type 1. Type 2 diabetes is obesity-driven, insulin-resistant, and carries acanthosis nigricans and a type 2 family history, so a lean child with mild, stable hyperglycaemia and a dominant pedigree is not type 2. [1] [5]
Points to monogenic
- Autosomal-dominant family history
- Islet autoantibodies negative
- C-peptide preserved beyond honeymoon
- Onset under six months, or mild stable hyperglycaemia
Points to type 1
- Acute osmotic or ketoacidotic onset
- Islet autoantibodies positive
- C-peptide falls over time
- No dominant diabetes pedigree
Points to type 2
- Obese adolescent with acanthosis nigricans
- Insulin-resistant phenotype
- Strong type 2 family history
- Higher C-peptide and insidious onset
The neonatal differential includes the secondary causes of hyperglycaemia in a sick neonate — sepsis, steroid therapy, and the stress of critical illness — but persistent insulin-requiring hyperglycaemia beyond the acute episode, especially under six months, is neonatal diabetes and warrants genetic testing rather than attribution to stress alone. Transient stress hyperglycaemia resolves; monogenic neonatal diabetes persists. [7] [11]
Within monogenic diabetes itself, the differential is between the genes, and it is settled by molecular testing. The clinical pointers help direct the panel: a stable mild fasting glucose since birth favours GCK; a progressive adolescent onset with a dominant history favours HNF1A; renal cysts or pancreatic hypoplasia favour HNF1B; a neonatal onset with developmental delay favours a potassium-channel gene; and consanguinity with a syndromic picture raises the recessive genes such as EIF2AK3 and PDX1. [1] [10]
Clinical & Bedside Assessment
The bedside assessment of a neonate with diabetes is a sick-infant assessment first. Secure the airway, breathing and circulation, treat dehydration and electrolyte disturbance, and start insulin to control the hyperglycaemia while the diagnostic process proceeds, because a sustained high glucose in a developing brain is itself harmful. The gene is found in due course, not at the bedside, and the initial priority is metabolic stability. [1] [11]
The general examination searches for the syndromic clues that point to specific genes. Plot the growth, because intrauterine growth restriction is common in neonatal diabetes from foetal insulin deficiency. Examine the musculoskeletal system for the epiphyseal dysplasia of Wolcott-Rallison syndrome, the abdomen for hepatosplenomegaly or recurrent hepatic dysfunction, and the neurology for the developmental delay, hypotonia and seizures of DEND syndrome. Renal anomalies on imaging point to HNF1B-MODY. [10] [12]
In the older child with suspected MODY, the assessment is directed at the discriminators. Confirm the body mass index and look for acanthosis nigricans, which argue for type 2. Examine for the pigmentation of Addison disease and the goitre of autoimmune thyroiditis, which would steer the work-up toward autoimmune type 1 or a polyglandular syndrome rather than monogenic disease. Measure the blood pressure and dip the urine, because renal involvement in HNF1B-MODY may be the presenting feature. [2] [5]
A careful three-generation family history is the single most useful bedside tool. Ask specifically about relatives diagnosed young, relatives on insulin whose antibody status is unknown, relatives with renal cysts or chronic kidney disease of unclear cause, and relatives with recurrent pancreatitis. Drawing the pedigree often makes the dominant pattern obvious where the narrative had not, and it identifies relatives who themselves warrant cascade testing once the proband's gene is found. [1] [6]
Investigations
The investigations proceed in two steps: first confirm that the diabetes is not autoimmune, then confirm the gene. Measure the islet autoantibodies — glutamic acid decarboxylase, insulinoma-associated antigen 2, insulin and zinc transporter 8 — because a negative antibody panel in a child with retained endogenous insulin is the opening signal for monogenic disease. A C-peptide level drawn with a simultaneous glucose measures residual beta-cell function, and a C-peptide that persists years after diagnosis, beyond any honeymoon, argues strongly against type 1. [1] [6]
A population study of a biomarker-based screening pathway showed that combining a young age at onset, a negative antibody status and a measurable C-peptide identifies the children most likely to carry a monogenic mutation, focusing expensive genetic testing on the highest-yield candidates. Urine C-peptide creatinine ratio is now used as a convenient surrogate for stimulated C-peptide in screening, sparing the child a fasting blood draw. [6]
Genetic testing is the diagnostic standard. A targeted next-generation sequencing panel covering the common MODY and neonatal diabetes genes is the usual first test, with whole-exome or whole-genome sequencing reserved for cases where the panel is unrevealing or the phenotype is syndromic. Testing should be done through a specialist monogenic diabetes service with access to genetic counselling, because the result changes not only the proband's treatment but the testing and counselling offered to the whole family. [1] [2]
For the child presenting in the neonatal period, the first-hour metabolic bundle also matters while the gene is pending: a venous gas, blood glucose, electrolytes, calcium, magnesium, ketones, a full blood count, and a septic screen to exclude the commoner neonatal emergencies. HbA1c at presentation quantifies the chronicity, and thyroid function testing screens for the associated autoimmune thyroid disease that, while uncommon in strictly monogenic diabetes, is part of the routine diabetic work-up. [7] [11]
Where HNF1B-MODY is suspected, image the kidneys and the pancreas. Renal ultrasound identifies the cystic or hypoplastic dysplasia that often precedes the diabetes, and abdominal imaging may reveal pancreatic hypoplasia or atrophy. These findings, paired with the gene result, define the multi-system surveillance the child will need for life. [1] [10]
Management — Resuscitation
Resuscitation in neonatal diabetes is the resuscitation of a sick hyperglycaemic neonate, and it does not wait for the gene. Correct dehydration and electrolyte disturbance, treat any intercurrent sepsis, and start insulin to bring the glucose into a safe range, because the priority in the acute phase is metabolic stability and protection of the developing brain from sustained hyperglycaemia. The molecular result, when it arrives, redirects the long-term treatment. [1] [11]
Intravenous insulin by infusion is the usual acute modality in a neonate, titrated to a glucose target that avoids both hyperglycaemia and the hypoglycaemia that an immature brain tolerates poorly. A continuous intravenous insulin infusion at roughly 0.05 units per kilogram per hour, adjusted to response, is a common starting approach in specialist neonatal units, with transition to subcutaneous insulin as the child stabilises. [1]
The key resuscitative principle is that insulin is the bridge, not the destination, for a child who turns out to have a potassium-channel mutation. Once a KCNJ11 or ABCC8 activating mutation is confirmed, the planned move is to oral glibenclamide, which closes the channel directly and typically achieves better glycaemic control than insulin while also improving the neurological features. The resuscitation phase keeps the child safe; the molecular answer changes the maintenance therapy. [4] [10]
For a MODY child who is not acutely unwell, there is usually no resuscitation phase — the presentation is often incidental or elective. The exception is the HNF1A or HNF4A child who presents in ketoacidosis, who is managed with the standard paediatric diabetic ketoacidosis protocol with isotonic fluids, deferred insulin and potassium, as set out in the dedicated ketoacidosis leaf. The gene does not change the acute resuscitation, only the maintenance plan that follows. [1]
Management — Definitive & Stepwise
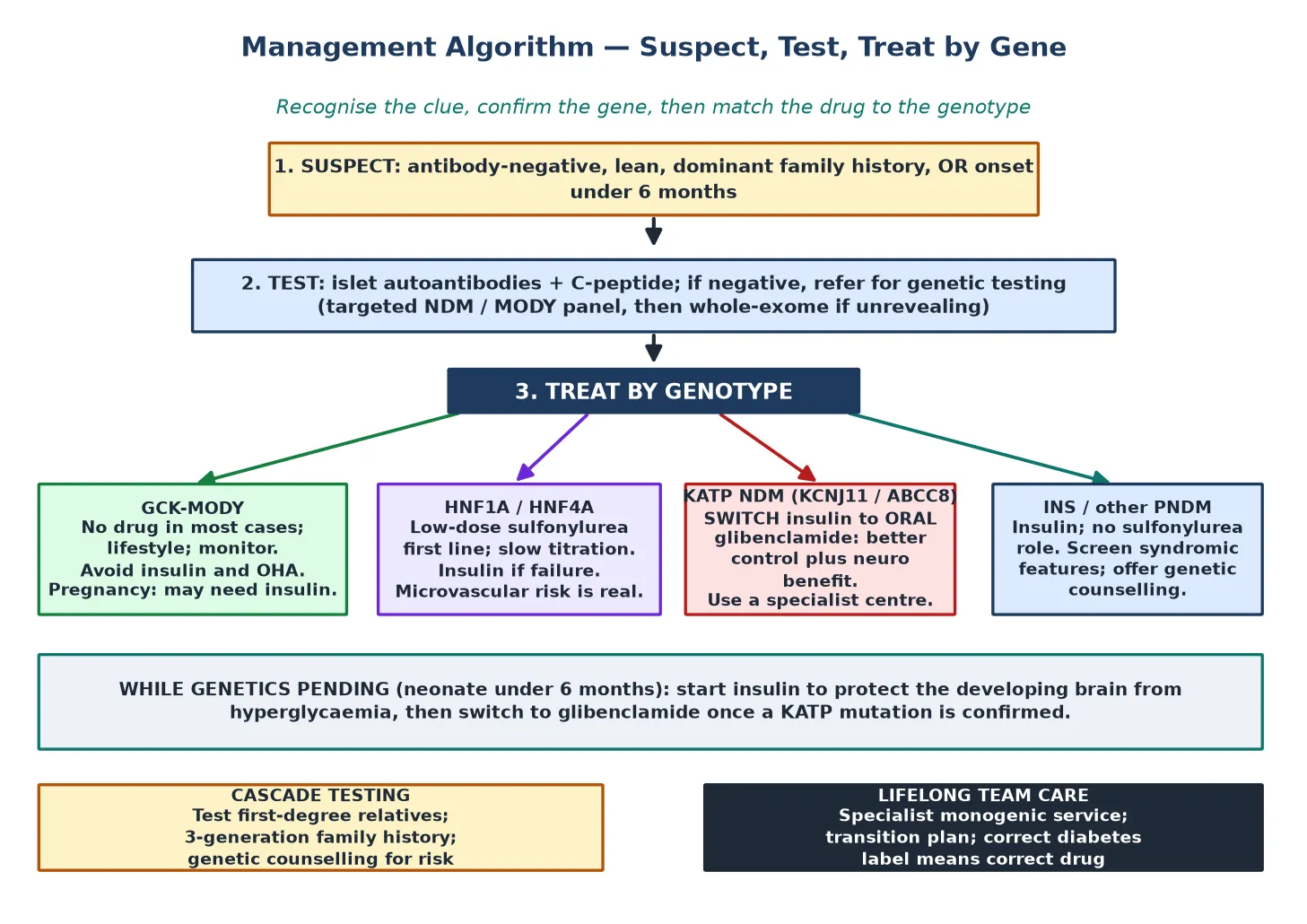
The definitive management of monogenic diabetes is set entirely by the gene, and the figure lays out the four treatment arms that follow from the four common mechanism groups. Confirm the molecular diagnosis, then match the drug to the genotype, then build the lifelong team care around it. [1]
Glucokinase-MODY is a raised glucose set-point, not a progressive disease, and most affected children need no pharmacotherapy at all. Microvascular complications are rare, and the risks of unnecessary insulin or oral hypoglycaemic agents — chiefly hypoglycaemia and a lifelong wrong label — outweigh any benefit. Treatment is reserved for pregnancy, where maternal hyperglycaemia affects foetal growth and insulin may be needed to optimise glycaemia, guided by the foetal abdominal circumference on ultrasound. [1] [8]
HNF1A-MODY and HNF4A-MODY are exquisitely sensitive to low-dose sulfonylureas, which are first-line. Start low and titrate slowly, because these patients respond to doses far below those used in type 2 diabetes and are prone to hypoglycaemia on over-treatment. Insulin is reserved for those who fail or cannot tolerate a sulfonylurea. Because the HNF forms carry a real microvascular risk over time, the glycaemic target is meaningful, and annual complication screening applies from adolescence as it does for any longstanding diabetes. [1] [2]
Potassium-channel neonatal diabetes is the transformative case. Once a KCNJ11 or ABCC8 activating mutation is confirmed, the child is switched from insulin to oral glibenclamide, which closes the channel directly and restores endogenous insulin release. The switch usually improves glycaemic control and, because the same channel operates in the brain, often improves the developmental and neurological features of DEND syndrome. The transfer is best managed through a specialist centre experienced in glibenclamide conversion, because the dose and the wean of insulin require careful coordination. [4] [10]
INS and other permanent neonatal diabetes that does not involve the potassium channel stays on insulin, because the beta cells are gone rather than silent, and there is no sulfonylurea role. These children need a subcutaneous insulin regimen individualised to age and feeding pattern, with the multidisciplinary support of a paediatric diabetes team. The syndromic forms also demand surveillance for their extra-pancreatic features — hepatic and skeletal disease in Wolcott-Rallison, exocrine function in PDX1, and renal and pancreatic imaging in HNF1B. [12] [10]
Across all subtypes, education, genetic counselling and family communication are as central as the drug. The family needs to understand the gene, its inheritance, the implications for relatives, and the rationale for the chosen treatment, delivered by a team that includes a paediatric endocrinologist, a diabetes nurse educator, a dietitian, and a clinical geneticist or genetic counsellor. The correct molecular label removes a wrong diagnosis as much as it adds a right one, and that correction is itself therapeutic. [1]
Specific Subtypes & Scenarios
Glucokinase-MODY is the subtype most often over-treated. The child is found to have a fasting glucose in the mild range, is labelled diabetic, and is started on a drug that adds hypoglycaemia to a condition that rarely causes complications. Recognising the stable, lifelong, mild fasting hyperglycaemia with a dominant family history, confirming the GCK mutation, and stepping back from pharmacotherapy is the management. Pregnancy is the one scenario where treatment is reconsidered, guided by foetal growth. [1] [8]
HNF1B-MODY is the multi-system scenario that demands a renal and pancreatic lens. The diabetes often emerges after the renal diagnosis, with hypoplastic or cystic kidney disease, chronic kidney disease of unclear cause, recurrent pancreatitis from pancreatic hypoplasia, or genitourinary anomalies. Imaging the kidneys and pancreas at diagnosis, and arranging longitudinal renal surveillance, is part of the management of the gene, not a separate problem. [10]
The potassium-channel neonatal diabetes with DEND syndrome is the scenario where treatment is most transformative. A child presenting with neonatal diabetes together with developmental delay, hypotonia or epilepsy may have a KCNJ11 or ABCC8 mutation in which the glibenclamide switch improves not only the diabetes but also the neurological features, because the channel defect is shared by brain and beta cell. Recognising the syndrome early and referring for glibenclamide conversion can change the developmental trajectory. [3] [4]
Transient neonatal diabetes is the scenario that is not as benign as the name suggests. The diabetes remits by around eighteen months, which can tempt a family and clinician to consider it resolved, but it frequently recurs in adolescence or adulthood and may require insulin again. Anticipatory counselling, ongoing monitoring through the remission, and a clear plan for recognising relapse are part of the management, because the gene persists even when the glucose normalises. [2] [11]
Wolcott-Rallison syndrome from biallelic EIF2AK3 mutations is the rare but severe scenario to hold in consanguineous families. It pairs neonatal or early-infantile diabetes with epiphyseal dysplasia, recurrent hepatic dysfunction, and a high risk of early mortality, and it demands a coordinated multi-system approach. PDX1 mutations illustrate the reverse: neonatal diabetes may occur without exocrine pancreatic insufficiency, reminding the clinician that the exocrine and endocrine pancreas can be dissociated even within a single gene. [12]
Complications & Pitfalls
The complications of monogenic diabetes are gene-specific. The HNF1A and HNF4A forms carry a real risk of retinopathy, nephropathy and neuropathy over time, so annual complication screening from adolescence applies as it does for any longstanding hyperglycaemia. HNF1B-MODY leads to progressive renal disease that may dominate the clinical course. The syndromic neonatal forms carry their own extra-pancreatic morbidity — skeletal, hepatic and neurological — that drives prognosis as much as the diabetes itself. [1] [10]
The commonest and most consequential pitfall is the missed diagnosis. A child mislabelled as type 1 receives decades of unnecessary insulin, with the burden of injections and the risk of hypoglycaemia, when an HNF1A or HNF4A mutation would have allowed a low-dose sulfonylurea. A child mislabelled as type 2 receives escalating oral agents and weight-focused advice that miss a single-gene cause. The countermeasure is the antibody and C-peptide screen in any child with an atypical diabetic course, and a low threshold for genetic referral. [5] [6]
The reverse pitfall is over-treatment of glucokinase-MODY. Placing a GCK child on insulin or an oral hypoglycaemic agent adds hypoglycaemia and a wrong lifelong label to a condition that rarely needs a drug, because the microvascular risk of glucokinase-MODY is low. Recognising the stable mild fasting hyperglycaemia, confirming the GCK mutation, and stepping back from pharmacotherapy is the correct, and often reassuring, management. [1] [8]
The neonatal pitfall is attributing early hyperglycaemia to stress and missing a potassium-channel mutation. A baby under six months with persistent insulin-requiring hyperglycaemia has neonatal diabetes until genetics says otherwise, and a confirmed KCNJ11 or ABCC8 mutation opens the option of a switch to oral glibenclamide with better glycaemic and neurological outcomes. Treating every neonatal diabetic as a future type 1 patient denies that opportunity. [7] [4]
Prognosis & Disposition
Prognosis in monogenic diabetes is governed by the gene. Glucokinase-MODY carries an excellent prognosis with a low complication rate and usually no need for treatment. The HNF1A and HNF4A forms carry a microvascular risk that is mitigated by good glycaemic control from diagnosis, achievable on a sulfonylurea in most cases. HNF1B-MODY prognosis is shaped by the renal disease as much as the diabetes. The potassium-channel neonatal diabetes, once switched to glibenclamide, often has markedly improved glycaemic and neurological outcomes compared with continued insulin. [1] [4]
The syndromic neonatal forms carry a more guarded prognosis. Wolcott-Rallison syndrome, with its recurrent hepatic failure and skeletal disease, is associated with significant early mortality. The prognosis of INS and other permanent neonatal diabetes on insulin is driven by glycaemic control and the absence of extra-pancreatic features. Transient neonatal diabetes remits but frequently recurs, so the prognosis is one of lifelong monitoring rather than cure. [10] [12]
Disposition is lifelong specialist care. Every child with confirmed monogenic diabetes should be managed with or shared with a specialist monogenic diabetes service, because the genotype-matched treatment, the cascade testing of relatives, the genetic counselling for reproductive risk, and the longitudinal surveillance of the syndromic features all exceed the scope of a general clinic. A structured transition to adult care in late adolescence preserves continuity, and a correct patient identifier protects against the wrong-diagnosis errors that recur across a lifetime of healthcare contacts. [1] [2]
Family communication is part of disposition. A molecular diagnosis in one family member changes the testing and counselling offered to parents, siblings, and future children, and identifying an affected relative can move them from insulin to a sulfonylurea, or from unnecessary treatment to none. The family, not only the proband, is the unit of care in monogenic diabetes. [6]
Special Populations
The neonate under six months is the population in which monogenic diabetes is most certain and most consequential. Every baby with diabetes in this window is presumed to have a monogenic cause, the acute priority is metabolic stability, and the molecular result redirects long-term treatment, often toward a switch from insulin to oral glibenclamide. The syndromic features — developmental delay, epilepsy, skeletal and hepatic disease — must be sought and managed alongside the diabetes. [7] [11]
The older child or adolescent previously labelled type 1 or type 2 is the population in which monogenic diabetes is most often missed. Re-testing antibodies and C-peptide and taking a three-generation family history in any atypical course uncovers the HNF1A and HNF4A MODY cases that hide inside the type 1 cohort, and the lean, antibody-negative children misdiagnosed as type 2. The benefit of reclassification is a simpler, safer treatment — a low-dose sulfonylurea, or no drug at all for glucokinase-MODY. [5] [6]
Consanguineous families carry a higher risk of the rarer recessive neonatal diabetes genes. Biallelic EIF2AK3, PDX1 or FOXP3 mutations produce syndromic neonatal or early-infantile diabetes with extra-pancreatic features that dominate the clinical picture, and a careful family history and dysmorphology examination direct the genetic panel. These families warrant genetic counselling for recurrence risk in future pregnancies. [12] [10]
Remote, Indigenous and migrant populations face barriers of access to genetic testing and specialist monogenic services that the disease itself does not impose. Telehealth links to specialist centres, locally delivered diabetes education, and culturally safe family communication narrow the gap, because the molecular diagnosis and the genotype-matched treatment should not depend on geography. The benefit of a correct label — fewer injections, less hypoglycaemia, the right surveillance — is greatest where access has been hardest. [1]
Evidence, Guidelines & Regional Differences
The guideline base for monogenic diabetes in children is the ISPAD Clinical Practice Consensus Guidelines on the diagnosis and management of monogenic diabetes, with the 2018 update and its 2014 predecessor setting the international standard for suspicion, genetic testing and genotype-matched treatment. National paediatric diabetes bodies broadly align, and specialist monogenic diabetes centres publish practical referral pathways that translate the guidelines into local practice. [1] [2]
The molecular evidence is anchored by the discovery that activating mutations in the KCNJ11 gene encoding the Kir6.2 subunit cause permanent neonatal diabetes, and by the demonstration that patients with these mutations can be switched from insulin to oral sulfonylureas. The demonstration that KCNJ11 mutations are a common cause of diabetes diagnosed in the first six months, with the phenotype set by genotype, established the six-month rule and the centrality of genetic testing in neonatal diabetes. [3] [4] [7]
The epidemiological evidence comes from the SEARCH study, which documented the prevalence and the clinical mislabelling of HNF1A, HNF4A and glucokinase MODY in youth, and from population work on biomarker-based screening pathways that combine young onset, negative antibodies and a measurable C-peptide to target genetic testing. The Italian national survey tracking two decades of neonatal diabetes showed the expanding genetic and phenotypic spectrum as testing widened. [5] [6] [11]
The subtype evidence covers the spectrum: heterozygous ABCC8 mutations as a cause of MODY, the role of activating ABCC8 and KCNJ11 mutations in permanent neonatal diabetes, the recognition of GCK-MODY in neonatal hyperglycaemia, and the demonstration that biallelic PDX1 mutations cause neonatal diabetes sometimes without exocrine insufficiency. Together these define the breadth of the monogenic landscape that a clinical genetic panel must cover. [8] [9] [10] [12]
Current controversies and regional differences turn on access. Genetic testing availability, funding pathways for the tests and for glibenclamide conversion, and the organisation of specialist monogenic services vary between and within countries, which affects how promptly a child is correctly labelled and switched to genotype-matched treatment. The move toward wider biomarker screening of the existing type 1 and type 2 cohorts, and toward whole-genome sequencing for unrevealing panel-negative cases, is narrowing the diagnostic gap but is unevenly distributed. [1] [6]
Exam Pearls
The one-sentence exam answer for monogenic diabetes is that a single-gene beta-cell defect masquerading as type 1 or type 2 diabetes is identified by young onset, negative antibodies, a preserved C-peptide and a dominant family history, confirmed by genetic testing, and treated by genotype — no drug for GCK, a low-dose sulfonylurea for HNF1A and HNF4A, and a switch to oral glibenclamide for potassium-channel neonatal diabetes. [1]
State the frequently-misremembered facts: diabetes under six months is almost never autoimmune type 1; a GCK-MODY child usually needs no drug; HNF1A and HNF4A MODY are sulfonylurea-sensitive and should not be defaulted to insulin; and a KCNJ11 or ABCC8 neonatal diabetic can usually be switched from insulin to oral glibenclamide for better glucose and neurological outcomes. Transient neonatal diabetes remits but frequently recurs, so it is not cured. [4] [7]
Give the high-yield triggers for genetic testing: onset under six months; a dominant family history across three generations; antibody-negative diabetes with a C-peptide that persists beyond the honeymoon; a lean child with mild, stable hyperglycaemia; and renal cysts or pancreatic hypoplasia pointing to HNF1B-MODY. Any one of these warrants a referral for molecular testing. [1] [6]
Give the high-yield lesion-sign pairings: a parent and child both on insulin with negative antibodies is HNF1A or HNF4A MODY, not type 1; mild stable fasting hyperglycaemia from birth is glucokinase-MODY and usually needs no drug; neonatal diabetes with developmental delay and epilepsy is a potassium-channel DEND syndrome amenable to glibenclamide; and neonatal diabetes with epiphyseal dysplasia and hepatic dysfunction in a consanguineous family is Wolcott-Rallison syndrome. [3] [10] [12]
References
- [1]Hattersley AT; Greeley SAW; Polak M; et al ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes, 2018.PMID 30225972
- [2]Rubio-Cabezas O; Hattersley AT; Njølstad PR; et al ISPAD Clinical Practice Consensus Guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes, 2014.PMID 25182307
- [3]Gloyn AL; Pearson ER; Antcliff JF; et al Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med, 2004.PMID 15115830
- [4]Pearson ER; Flechtner I; Njølstad PR; et al Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med, 2006.PMID 16885550
- [5]Pihoker C; Gilliam LK; Ellard S; et al Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab, 2013.PMID 23771925
- [6]Shields BM; Hicks S; Shepherd MH; et al Population-Based Assessment of a Biomarker-Based Screening Pathway to Aid Diagnosis of Monogenic Diabetes in Young-Onset Patients. Diabetes Care, 2017.PMID 28701371
- [7]Flanagan SE; Edghill EL; Gloyn AL; et al Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia, 2006.PMID 16609879
- [8]Hughes AE; Maguiness S; Balci S; et al Identification of GCK-maturity-onset diabetes of the young in cases of neonatal hyperglycemia: A case series and review of clinical features. Pediatr Diabetes, 2021.PMID 34085361
- [9]Bowman P; Flanagan SE; Edghill EL; et al Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia, 2012.PMID 21989597
- [10]Edghill EL; Bingham C; Ellard S; Hattersley AT Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev Endocr Metab Disord, 2010.PMID 20922570
- [11]Rapini N; Martinez TF; Bizzarri C; et al The Changing Landscape of Neonatal Diabetes Mellitus in Italy Between 2003 and 2022. J Clin Endocrinol Metab, 2024.PMID 38408297
- [12]De Franco E; Flanagan SE; Houghton JAL; et al Biallelic PDX1 (insulin promoter factor 1) mutations causing neonatal diabetes without exocrine pancreatic insufficiency. Diabet Med, 2013.PMID 23320570