Paeds · endocrinology-diabetes-and-growth
Phaeochromocytoma and endocrine hypertension
Also known as Phaeochromocytoma · Paraganglioma · Catecholamine-secreting tumour · Monogenic hypertension · Low-renin hypertension · Endocrine hypertension · Mineralocorticoid hypertension
Fellowship guide to phaeochromocytoma, paraganglioma and the endocrine causes of hypertension in children: the metanephrines-not-catecholamines diagnostic rule, the high hereditary fraction that makes every child a genetics patient, the alpha-before-beta preoperative trap, and the renin–aldosterone fork that sorts the monogenic mineralocorticoid causes of low-renin hypertension.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The single idea that organises the whole topic is that endocrine hypertension in children splits into two mechanistic families: catecholamine-driven disease (phaeochromocytoma and paraganglioma) and mineralocorticoid-driven disease (the monogenic low-renin causes). The first is caught by metanephrines and fixed by alpha-blockade then surgery; the second is caught by a renin–aldosterone–potassium panel and fixed by matching the drug to the gene. [1] [11]
This page covers the recognition and management of phaeochromocytoma and paraganglioma in children, then turns to the broader endocrine causes of hypertension: the monogenic mineralocorticoid syndromes (Liddle, apparent mineralocorticoid excess, eleven- and seventeen-hydroxylase deficiency, Gordon syndrome), primary aldosteronism, and cortisol and thyroid excess. It links to the Cushing and congenital adrenal hyperplasia leaves for their dedicated pathways rather than repeating them. [2] [12]
Overview & Definition
Phaeochromocytoma and paraganglioma are tumours of the chromaffin cell, the neuroendocrine cell that stores and releases catecholamines. An adrenal medullary lesion is a phaeochromocytoma; an identical lesion arising in the extra-adrenal paraganglia — the organ of Zückerkandl, the head and neck, the thorax — is a paraganglioma. Both leak metanephrines and both can cause the same catecholamine storm. [1]
What sets the paediatric disease apart is its hereditary burden and its behaviour. Children more often present with sustained hypertension rather than the classic adult episodic spells, their tumours are more often bilateral, multifocal and extra-adrenal, and roughly four in ten carry a germline mutation. The old teaching that phaeochromocytomas follow a "rule of tens" — ten percent bilateral, malignant, extra-adrenal and familial — describes adult disease and badly undercalls the heritable fraction in children. [2] [5]
Endocrine hypertension is the broader umbrella for any hormone-driven rise in blood pressure. It includes the catecholamine tumours, the mineralocorticoid-excess syndromes (whether monogenic, aldosterone-driven or cortisol-driven), and hypertension from thyroid or parathyroid excess. The first step in any hypertensive child is therefore endocrine: measure metanephrines for the catecholamine family, and renin with aldosterone for the mineralocorticoid family. [10] [12]
Classification
Sort the disease by the hormone axis driving the blood pressure, because the axis dictates both the test and the treatment. The figure below splits endocrine hypertension into catecholamine-mediated phaeochromocytoma and paraganglioma, mineralocorticoid-excess syndromes, and glucocorticoid or thyroid causes. [1]
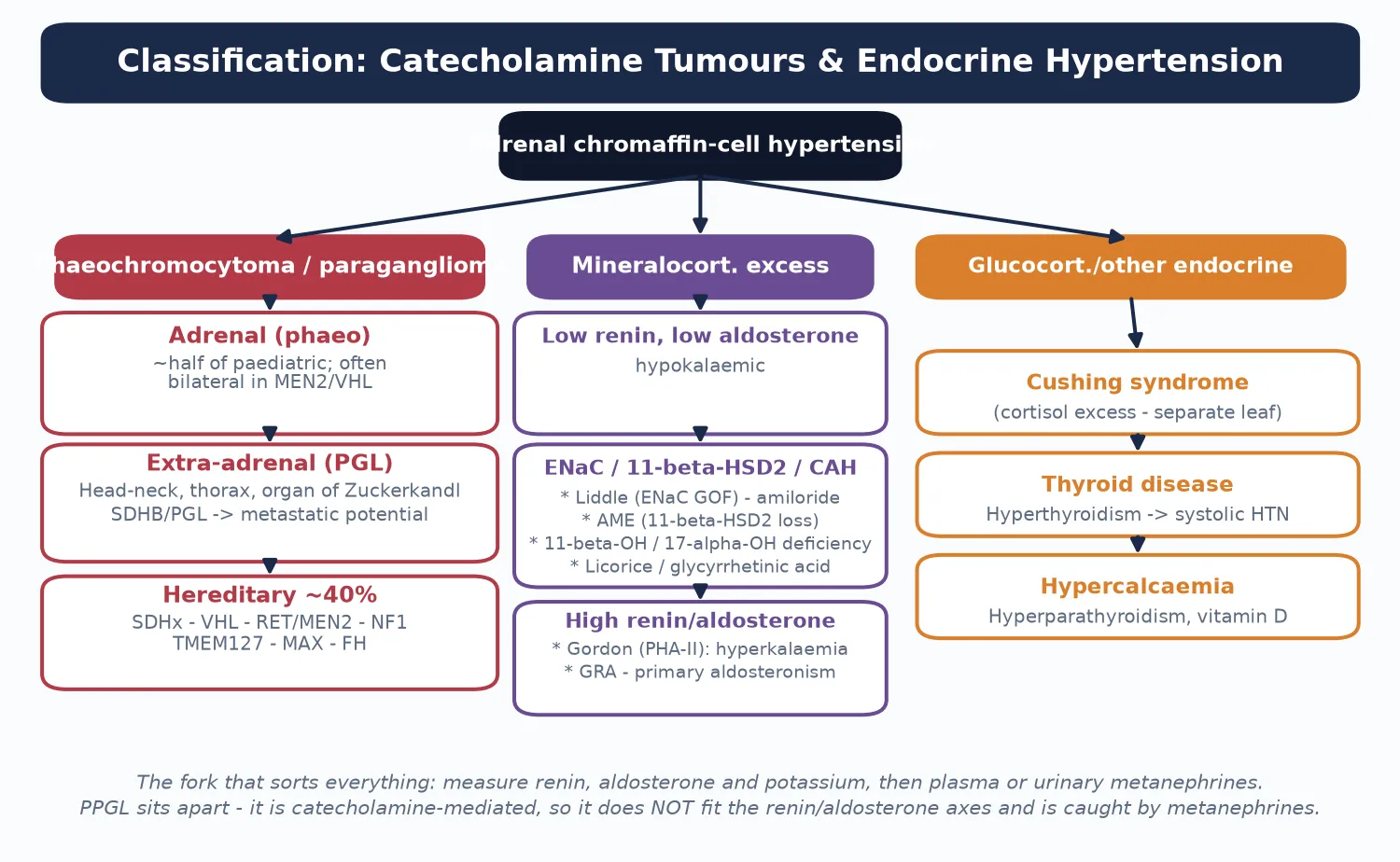
Catecholamine tumour (PPGL)
- Phaeochromocytoma (adrenal) or paraganglioma (extra-adrenal)
- Test: plasma free metanephrines or 24-h urine fractionated metanephrines
- Hereditary in ~40%: SDHx, VHL, RET (MEN2), NF1, TMEM127, MAX, FH
- Treat: alpha-blockade then surgery — never beta first
Low-renin, low-aldosterone
- Hypokalaemic, suppressed renin AND suppressed aldosterone
- Liddle syndrome (ENaC gain-of-function) — amiloride-responsive
- Apparent mineralocorticoid excess (11-beta-HSD2 loss)
- 11-beta- and 17-alpha-hydroxylase deficiency; licorice
High-renin or high-aldosterone
- Gordon syndrome (pseudohypoaldosteronism II) — hyperkalaemia
- Glucocorticoid-remediable aldosteronism (familial hyperaldosteronism I)
- Primary aldosteronism — adenoma or bilateral hyperplasia
- Renovascular disease (low-renin is rare here, but renin is high)
The renin and aldosterone pair does the sorting. A low renin with a low aldosterone points to a mineralocorticoid receptor being driven from below the adrenal — sodium retention at the distal tubule independent of aldosterone — which is the signature of Liddle, apparent mineralocorticoid excess and the hypertensive forms of congenital adrenal hyperplasia. A high aldosterone with a suppressed renin points to primary aldosteronism, while a high renin with a high aldosterone points to Gordon syndrome or renovascular disease. [11] [12]
Epidemiology & Risk Factors
Phaeochromocytoma and paraganglioma are rare in children, but they account for a notable minority of secondary hypertension in paediatric referral series. The paediatric incidence is far below the adult figure, and presentation clusters in later childhood and adolescence, though hereditary forms can declare themselves in infancy. [5] [6]
The dominant risk factor is heredity. Around forty percent of paediatric phaeochromocytomas and paragangliomas carry a pathogenic germline mutation, a fraction that dwarfs the ten percent familial figure inherited from adult teaching. The succinate dehydrogenase subunit genes (SDHB, SDHD, SDHC, SDHA and SDHAF2) are the commonest culprits; von Hippel–Lindau disease (VHL), multiple endocrine neoplasia type 2 (RET) and neurofibromatosis type 1 (NF1) account for most of the rest. A family history of early-onset endocrine tumours, sudden death or known syndromic features should lower the threshold to test. [2] [7]
The reason heredity looms so large in children is biological. Several of the susceptibility genes — the SDHx cluster — are tumour suppressors whose loss drives chromaffin-cell tumours early and often multifocally, so a child carrying an SDHB or SDHD mutation is on a lifelong surveillance pathway from the time the mutation is found. Carrying that expectation into the first consultation reframes the disease from a single resectable mass to an inherited cancer-predisposition syndrome. [7] [8]
Pathophysiology
To see why a catecholamine tumour behaves the way it does, picture the chromaffin cell as a leaky pressure cooker. Inside the tumour cell, catecholamines are continually being broken down to metanephrines — noradrenaline to normetanephrine, adrenaline to metanephrine — and these metabolites leak into the bloodstream continuously, whether or not the tumour releases a burst of catecholamine. That single fact is why metanephrines are the test and adrenaline is not. [3]
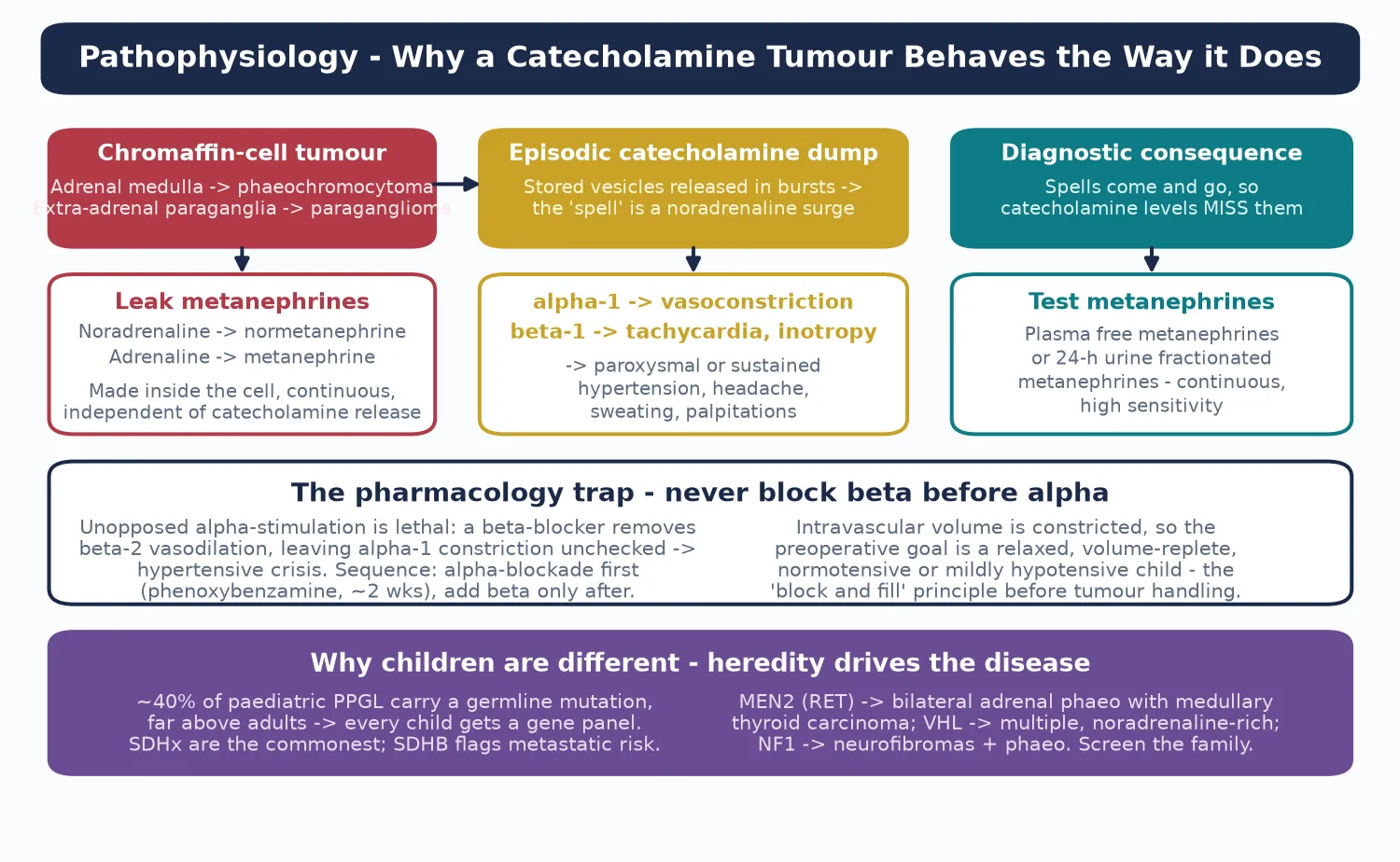
Two features of this loop matter at the bedside. First, catecholamine release is episodic, so a catecholamine level drawn between spells reads normal and misses the tumour; the continuously leaking metanephrines never do. Second, noradrenaline hits the alpha-one receptor to vasoconstrict, driving the hypertension, headache and pallor, while adrenaline hits the beta-one receptor to drive tachycardia and palpitations — and blocking beta before alpha leaves alpha-one unopposed and is lethal. [1] [3]
The hereditary causes share a theme: a defective oxygen-sensing or growth-control pathway that lets chromaffin cells proliferate. Loss of succinate dehydrogenase (the SDHx cluster) deranges cellular metabolism and stabilises hypoxia-inducible factors; VHL disease does the same through the von Hippel–Lindau protein; RET gain-of-function in MEN2 drives medullary thyroid carcinoma alongside bilateral phaeochromocytoma. Knowing the gene predicts the behaviour: SDHB marks extra-adrenal, metastatic paraganglioma, while MEN2 marks bilateral adrenal tumours with medullary thyroid cancer. [7] [8]
For the mineralocorticoid causes the pathophysiology turns on the distal nephron. Liddle syndrome is a gain-of-function mutation of the epithelial sodium channel (ENaC) that swallows sodium independent of aldosterone, dragging water with it and pouring out potassium; aldosterone and renin both fall. Apparent mineralocorticoid excess is a loss of the enzyme (11-beta-hydroxysteroid dehydrogenase type 2) that normally shields the mineralocorticoid receptor from cortisol, so cortisol floods the receptor. The hydroxylase-deficiency forms of congenital adrenal hyperplasia shunt steroid precursors into mineralocorticoid pathways. [11] [12]
Clinical Presentation
A child with a catecholamine tumour usually presents with hypertension and the spells that make adults famous for the disease, though in children the blood pressure is more often sustained. The classic triad is severe headache, generalised sweating and palpitations, occurring together in paroxysms with pallor, anxiety and sometimes vomiting. Between spells the child may look well, which is exactly why the catecholamine level drawn in a quiet clinic reads falsely normal. [4] [6]
The hypertension itself is often the gateway. A child found to be hypertensive on a routine examination, or presenting with headache, visual disturbance or heart failure, may have an unrecognised phaeochromocytoma, and the combination of sustained hypertension with episodic spells and weight loss or sweating should trigger biochemical testing rather than reassurance. In hereditary syndromes the presentation may be the associated tumour — a medullary thyroid carcinoma in MEN2, retinal or central nervous system haemangioblastomas in VHL, neurofibromas in NF1. [2] [8]
The endocrine hypertension causes declare themselves on the electrolyte panel and blood pressure alone. A child with early-onset hypertension, hypokalaemia and a family history of early stroke or hypertension at a young age has a monogenic mineralocorticoid cause until proven otherwise — Liddle syndrome is the archetype. Hypertension with hyperkalaemia and metabolic acidosis points to Gordon syndrome. A virilised hypertensive infant girl or under-virilised hypertensive infant boy points to eleven-beta- or seventeen-alpha-hydroxylase deficiency. [11] [12]
Differential Diagnosis
The differential turns on separating a catecholamine tumour from the other causes of hypertension in a child, and the biochemistry does most of the work. Essential (primary) hypertension is the commonest cause overall, especially in older obese adolescents, but it lacks the spells, the weight loss and the striking biochemical signal. Renovascular disease — fibromuscular dysplasia, neurofibromatosis-associated renal artery stenosis, vasculitis — causes high-renin hypertension with abdominal bruits and can mimic endocrine disease. [10]
Points to a catecholamine tumour
- Paroxysmal or sustained hypertension with spells
- Headache, sweating and palpitations as a triad
- Markedly raised plasma or urinary metanephrines
- Adrenal or extra-adrenal mass on imaging
- Family history of SDHx, VHL, MEN2 or NF1
Points to a mimic
- Essential hypertension: obese adolescent, no spells, normal metanephrines
- Renovascular disease: high renin, abdominal bruit, asymmetric kidneys
- Coarctation: upper-limb hypertension, radio-femoral delay, weak femoral pulses
- Renal parenchymal disease: raised creatinine, proteinuria, abnormal ultrasound
- Drug or substance effect: sympathomimetics, steroids, cocaine
Coarctation of the aorta is the non-endocrine cause that every candidate must mention, because it presents with hypertension and is excluded at the bedside with four-limb blood pressures. A monogenic mineralocorticoid cause enters the differential once renin and aldosterone return low, and the electrolyte and phenotype clues — hypokalaemia, virilisation, hyperkalaemia — redirect the work-up to the specific gene. Exogenous or iatrogenic causes — sympathomimetic decongestants, oral contraceptives, glucocorticoids, recreational stimulants — are caught by a careful drug history. [10] [11]
Clinical & Bedside Assessment
The focused assessment of a hypertensive child with suspected endocrine disease rests on measuring the blood pressure properly, plotting it, and reading the signs that redirect the work-up. Use an appropriate cuff and record four-limb pressures to exclude coarctation, and stage the blood pressure against normative paediatric thresholds — hypertension in a child is defined by percentile, not by an adult cut-off. [10]
Examine for the syndromic stigmata that flag a hereditary cause. café-au-lait patches and neurofibromas point to NF1; a retinal or central nervous system lesion history points to VHL; a thyroid mass with a marfanoid habitus and mucosal neuromas points to MEN2B; lentigines and atrial myxomas point to Carney complex (rarely associated with phaeochromocytoma). Measure the weight — weight loss despite a good appetite is a catecholamine clue — and look for proximal myopathy and the pallor of vasoconstriction. [2] [8]
The mineralocorticoid causes are read off the phenotype and electrolytes at the bedside. A virilised hypertensive infant raises eleven-beta-hydroxylase deficiency; an under-virilised hypertensive infant with sexual infantilism raises seventeen-alpha-hydroxylase deficiency; a hypokalaemic hypertensive child with a striking family history raises Liddle syndrome or apparent mineralocorticoid excess. Ask about licorice and chewing tobacco, which inhibit 11-beta-hydroxysteroid dehydrogenase type 2 and mimic apparent mineralocorticoid excess. [11] [12]
Investigations
The biochemical work-up proceeds in two streams that run in parallel: one for the catecholamine family and one for the mineralocorticoid family. For suspected phaeochromocytoma, measure plasma free metanephrines or a twenty-four-hour urine fractionated metanephrines collection — both are the recommended first-line tests because the tumour leaks these continuously. A clearly normal result excludes disease; a clearly raised result demands imaging. [1] [3]
Plasma free metanephrines
- Highest sensitivity — tumour leaks metanephrines continuously
- Preferred when clinical suspicion is high
- Falsely raised by caffeine, paracetamol, tricyclics, decongestants
- Draw supine, rested, off interfering drugs
24-h urine fractionated metanephrines
- Strong specificity, lower false-positive rate
- Useful for borderline plasma results and for screening
- Requires a complete collection — check creatinine
- Add urinary catecholamines for a fuller picture
Renin and aldosterone
- Sorts the mineralocorticoid arm of endocrine hypertension
- Low renin + low aldosterone = Liddle, AME, hydroxylase deficiency
- Low renin + high aldosterone = primary aldosteronism
- High renin + high aldosterone = Gordon syndrome, renovascular
Once the metanephrines are raised, localise the tumour. Computed tomography or magnetic resonance imaging of the abdomen and pelvis is first-line, because most tumours lie in the adrenal gland or the organ of Zückerkandl; MRI avoids radiation, which matters in children. Functional imaging then characterises and stages: iodine-123 meta-iodobenzylguanidine scintigraphy, and increasingly gallium-68 DOTATATE positron-emission tomography, which is more sensitive for metastatic and hereditary disease. [1] [5]
Genetic testing is not optional in a child. Every paediatric phaeochromocytoma and paraganglioma earns a hereditary gene panel covering the SDHx cluster, VHL, RET, NF1, TMEM127, MAX and FH, because identifying the mutation predicts tumour behaviour, sets the surveillance schedule, and enables cascade testing of relatives. An SDHB result converts an apparently localised paraganglioma into a malignancy-surveillance diagnosis. [2] [7]
For the mineralocorticoid causes, the renin–aldosterone–potassium panel is the gateway, followed by gene testing for the suspected syndrome. Liddle is confirmed by SCNN1B or SCNN1G sequencing; apparent mineralocorticoid excess by HSD11B2 sequencing; the hydroxylase deficiencies by CYP11B1 or CYP17A1 testing with the steroid precursor profile; Gordon syndrome by WNK1, WNK4, KLHL3 or CUL3 testing. Primary aldosteronism is confirmed by a suppressed renin with a high aldosterone, then lateralised with adrenal venous sampling in older children. [11] [12]
Management — Resuscitation
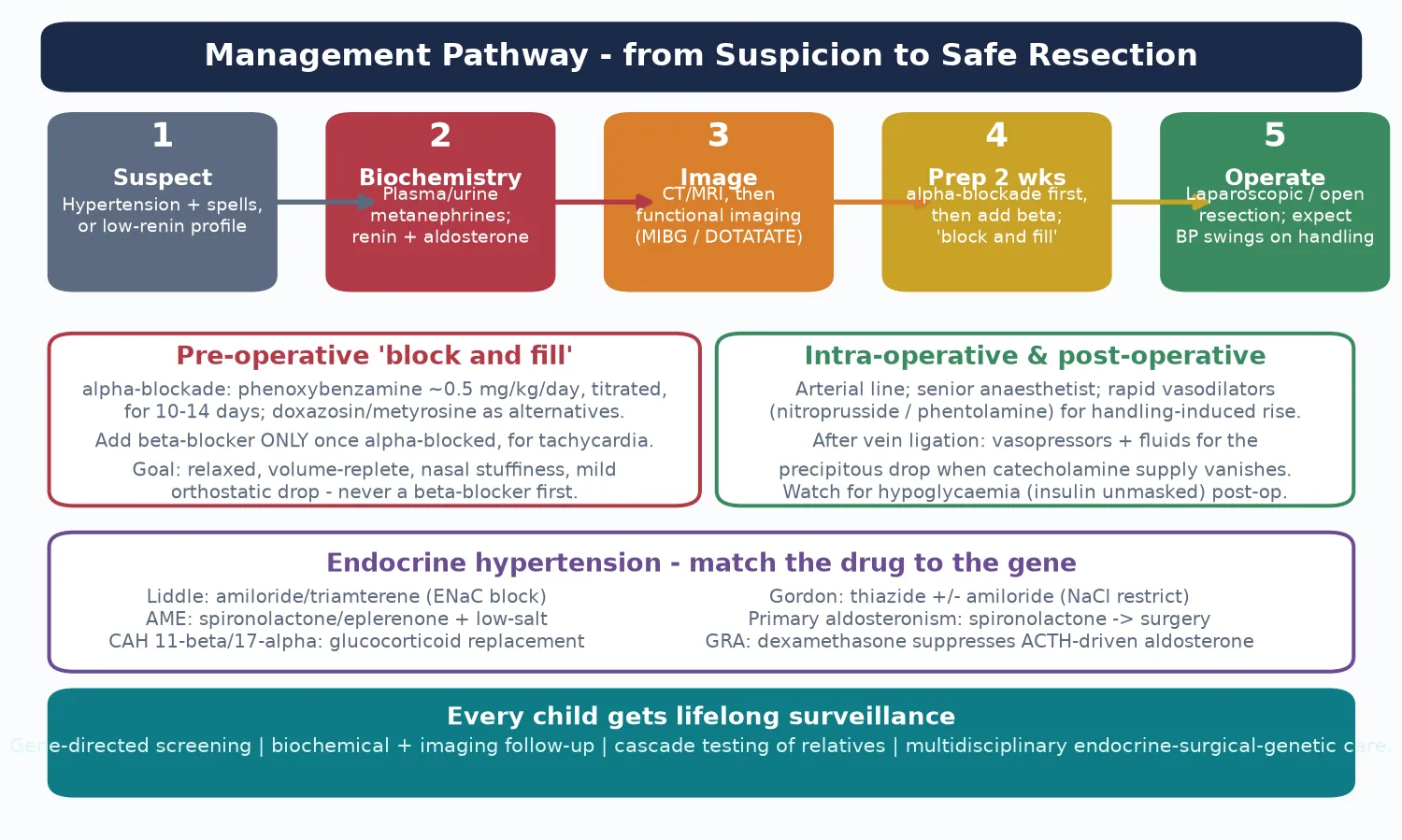
Phaeochromocytoma becomes a resuscitation emergency in two situations: the hypertensive crisis of an unblocked tumour (spontaneous or provoked by biopsy, anaesthesia or a beta-blocker), and the hypotension that follows tumour removal. Both are managed by a team that understands catecholamine pharmacology, and both are reasons to prepare the child meticulously rather than rush to theatre. [1]
A hypertensive crisis is treated with a rapid-acting intravenous vasodilator such as sodium nitroprusside or phentolamine, titrated to the blood pressure through an arterial line, with a senior intensivist and anaesthetist present. Do not reach for a beta-blocker to control the tachycardia until the alpha-receptors are blocked, because doing so deepens the crisis. Control the blood pressure first, expand the intravascular volume, and only then address the rhythm. [1] [5]
The opposite emergency — the hypotension after the adrenal vein is ligated — happens because the chronic vasoconstriction is suddenly lifted and the intravascular space is volume-contracted. The treatment is intravenous fluid and a vasopressor such as noradrenaline, guided by the arterial line, alongside vigilant monitoring for hypoglycaemia, which appears as the catecholamine-induced insulin suppression is lifted. Expect it, prepare for it, and the postoperative course is smooth. [1] [5]
Management — Definitive & Stepwise
Definitive treatment is surgical resection, but the operation begins two weeks earlier with preoperative alpha-blockade. Phenoxybenzamine, a non-competitive, irreversible alpha-blocker, is started at around 0.5 mg per kilogram per day in divided doses and titrated over ten to fourteen days until the child is normotensive or mildly hypotensive with an orthostatic drop and nasal stuffiness — the "block and fill" end-point. [1] [2]
A beta-blocker is added only after the child is fully alpha-blocked, to control reflex tachycardia — never before. Laparoscopic adrenalectomy is the standard for adrenal tumours in expert paediatric surgical hands, with conversion to open for large or extra-adrenal paragangliomas. The anaesthetic team expects wide blood-pressure swings on tumour handling, with nitroprusside or phentolamine ready for surges and vasopressors ready for the post-ligation drop. [1] [5]
For the mineralocorticoid causes, definitive treatment matches the drug to the gene. Liddle syndrome responds to the epithelial sodium channel blockers amiloride or triamterene, which correct the hypertension and the hypokalaemia directly at the channel that the mutation drives. Apparent mineralocorticoid excess and the hydroxylase-deficiency forms respond to glucocorticoid replacement or mineralocorticoid-receptor blockade; Gordon syndrome responds to thiazide diuretics with salt restriction. [11] [12]
Primary aldosteronism is managed medically with spironolactone or eplerenone, with unilateral adrenalectomy reserved for a clearly lateralised aldosterone-producing adenoma after adrenal venous sampling. Glucocorticoid-remediable aldosteronism is suppressed by low-dose dexamethasone. The point is that each monogenic cause has a rational, gene-directed first-line therapy, which is why identifying the mutation is therapeutic, not merely diagnostic. [9] [11]
Specific Subtypes & Scenarios
Phaeochromocytoma in MEN2 is the archetype of an adrenal hereditary tumour. RET gain-of-function drives medullary thyroid carcinoma in near-all carriers and bilateral adrenal phaeochromocytoma in a substantial fraction, so a child with a thyroid mass and hypertension has MEN2 until proven otherwise. Prophylactic thyroidectomy and lifelong biochemical surveillance anchor the management, and the phaeochromocytoma is resected with cortical-sparing adrenalectomy where possible to avoid adrenal insufficiency. [8]
SDHB-associated paraganglioma is the malignancy scenario. Extra-adrenal paragangliomas carrying an SDHB mutation have a real propensity to metastasise, so they are staged and surveyed as neuroendocrine malignancies with functional imaging. A child found to carry SDHB enters lifelong surveillance from diagnosis, and a metastatic deposit may declare years after the primary resection, which is why follow-up is indefinite rather than fixed. [2] [7]
Von Hippel–Lindau disease produces multiple, noradrenaline-secreting tumours that are often bilateral and associated with haemangioblastomas and renal cell carcinoma. Neurofibromatosis type 1 produces neurofibromas and a smaller lifetime risk of phaeochromocytoma. The family history and the syndromic stigmata guide both the surveillance schedule and the choice of imaging modality, because functional imaging differs by genotype. [5] [7]
Monogenic mineralocorticoid hypertension is the scenario the short case tests. A hypertensive child with hypokalaemia, a suppressed renin and aldosterone, and a striking family history of early-onset hypertension and stroke has Liddle syndrome until the gene test proves otherwise; the bedside win is knowing that amiloride, not spironolactone, is the rational therapy because the problem is the channel, not the receptor. A hypertensive virilised infant has eleven-beta-hydroxylase deficiency. [11] [12]
Complications & Pitfalls
The untreated catecholamine tumour carries a heavy burden: hypertensive heart failure with cardiomyopathy, hypertensive encephalopathy and stroke, and the metabolic consequences of chronic catecholamine excess. Many of these are reversible with resection, but a cardiomyopathy left too long may not recover, which is why early biochemical diagnosis is the single biggest modifiable prognostic factor. [5] [6]
The treatment carries its own pitfalls. Biopsy of an unblocked tumour can precipitate a fatal hypertensive crisis, so an adrenal mass in a child is never biopsied until phaeochromocytoma is biochemically excluded. A beta-blocker before an alpha-blocker is the classic lethal pharmacology error. Missing the hereditary mutation denies the child and the family a surveillance pathway and the chance to find a second tumour early. Each is an avoidable harm rooted in forgetting the disease's rules. [1] [2]
The mineralocorticoid causes have their own pitfalls. Treating Liddle syndrome with spironolactone is a common error that under-controls the blood pressure because the problem is the epithelial sodium channel, not the mineralocorticoid receptor — amiloride is the rational drug. Missing apparent mineralocorticoid excess because the history of licorice intake was not taken, and misattributing the hypertension of hydroxylase deficiency to essential hypertension in an undervirilised adolescent, are the diagnostic traps. [11] [12]
Prognosis & Disposition
With complete resection and expert perioperative care, the prognosis of a localised, non-metastatic phaeochromocytoma is excellent, and the hypertension usually resolves. Catch-up of the cardiomyopathy and the metabolic disturbance follows cure. The long-term battleground is the hereditary fraction: recurrence, a second primary tumour, or metastatic disease can declare years later, so surveillance is indefinite and gene-directed. [2] [7]
SDHB-associated and metastatic paraganglioma carry a guarded prognosis driven by tumour burden, resectability and response to therapy, including radiopharmaceutical agents such as iodine-131 MIBG. The monogenic mineralocorticoid causes, by contrast, carry an excellent prognosis once the correct gene-directed therapy is started — blood pressure is controlled, potassium normalises, and the family is offered cascade testing. Disposition is to a specialist paediatric endocrine service with adrenal-surgical, oncology and genetics links, and a planned transition to adult care. [7] [11]
Special Populations
Very young children with a catecholamine tumour are more likely than older children to carry a hereditary mutation and to have extra-adrenal or multifocal disease, so the threshold for a full gene panel and functional imaging is lower, not higher. The preoperative alpha-blockade and surgical principles are identical, but the anaesthetic and fluid management is more delicate in a small child. [2] [5]
The hereditary-syndrome child — MEN2, VHL, NF1, an SDHx carrier — is managed within a structured surveillance programme that coordinates endocrinology, genetics, oncology and the relevant organ specialist (thyroid for MEN2, ophthalmology and neurosurgery for VHL, dermatology for NF1). The family is offered cascade genetic testing, and the child carries a written surveillance plan into adult care. [7] [8]
Adolescents face the added burden of body-image and fertility concerns, adherence to a lifelong surveillance plan, and the transition to adult endocrine and genetics services. Indigenous, migrant and remote-dwelling families may face the logistics of accessing a specialist adrenal-surgical and genetics service, so a clear local pathway, telehealth support and a written emergency plan that travels with the child are essential, especially across the transition. [10]
Evidence, Guidelines & Regional Differences
The evidence base is anchored by the Endocrine Society clinical practice guideline on phaeochromocytoma and paraganglioma (Lenders and colleagues, 2014), which sets the metanephrines-first diagnostic standard and the alpha-before-beta preoperative rule. The 2024 international consensus on phaeochromocytoma and paraganglioma in children and adolescents (Casey and colleagues) brings the paediatric-specific recommendations together, and the Endocrine Society primary-aldosteronism guideline (Funder and colleagues, 2016) governs the high-aldosterone arm. [1] [9]
The paediatric-specific evidence draws on the cohort description of Barontini and colleagues, the reviews of Havekes and of Ganesh and colleagues, the hereditary genetics and surveillance guidelines of Muth and colleagues, and the MEN2B natural-history study of Castinetti and colleagues. The monogenic-hypertension references of New and of Garovic and colleagues anchor the mineralocorticoid arm, and Kapur and Baraco ground the evaluation of the hypertensive child. [4] [5]
Regional practice differences are modest because the Endocrine Society guidelines are adopted internationally, but access to functional imaging (gallium-68 DOTATATE), paediatric adrenal surgeons and rapid genetic panel testing varies between centres. In Australia and New Zealand, suspected paediatric phaeochromocytoma is referred to a tertiary paediatric endocrine service with a linked surgical, oncology and genetics team, reflecting the centralised expertise that the evidence supports. [2] [7]
The main controversies are the optimal duration and intensity of preoperative alpha-blockade, the role of cortical-sparing adrenalectomy in bilateral hereditary disease to preserve adrenal function, and the surveillance intensity after a seemingly complete resection of an SDHB-associated tumour. The evidence in these areas is limited in children and is individualised within a multidisciplinary team. [2] [9]
Exam Pearls
Hold one sentence for the viva: a hypertensive child with headache, sweating and palpitations has a catecholamine-secreting tumour until proven otherwise, and the diagnostic rule is to measure metanephrines — not catecholamines — because the tumour leaks the metabolite continuously while the hormone release is episodic. [1] [3]
State the frequently misremembered facts correctly. The preoperative sequence is alpha-blockade with phenoxybenzamine first, then a beta-blocker only after — never reversed, because unopposed alpha-one vasoconstriction is lethal. The hereditary fraction in children is about forty percent, not the ten percent of the outdated rule, so every child earns a gene panel. Plasma free metanephrines have the highest sensitivity; twenty-four-hour urine fractionated metanephrines the highest specificity. SDHB marks extra-adrenal, metastatic paraganglioma. [1] [7]
The high-yield pairings: a hypertensive child with spells and raised metanephrines is a phaeochromocytoma or paraganglioma; a hypertensive child with hypokalaemia and suppressed renin and aldosterone is Liddle syndrome (amiloride-responsive); a hypertensive virilised infant is eleven-beta-hydroxylase deficiency; a hypertensive under-virilised infant is seventeen-alpha-hydroxylase deficiency; a hypertensive child with hyperkalaemia is Gordon syndrome. These pairings do most of the diagnostic work in the short case. [11] [12]
References
- [1]Lenders JW; Duh QY; Eisenhofer G; et al Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab, 2014.PMID 24893135
- [2]Casey RT; Hendriks E; Deal C; et al International consensus statement on the diagnosis and management of phaeochromocytoma and paraganglioma in children and adolescents. Nat Rev Endocrinol, 2024.PMID 39147856
- [3]Lenders JW; Pacak K; Walther MM; et al Biochemical diagnosis of pheochromocytoma: which test is best? JAMA, 2002.PMID 11903030
- [4]Barontini M; Levin G; Sanso G Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann N Y Acad Sci, 2006.PMID 17102069
- [5]Havekes B; Romijn JA; Eisenhofer G; et al Update on pediatric pheochromocytoma. Pediatr Nephrol, 2009.PMID 18566838
- [6]Ganesh HK; Acharya SV; Goerge J; et al Pheochromocytoma in children and adolescents. Indian J Pediatr, 2009.PMID 20072855
- [7]Muth A; Crona J; Gimm O; et al Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J Intern Med, 2019.PMID 30536464
- [8]Castinetti F; Waguespack SG; Machens A; et al Natural history, treatment, and long-term follow up of patients with multiple endocrine neoplasia type 2B: an international, multicentre, retrospective study. Lancet Diabetes Endocrinol, 2019.PMID 30660595
- [9]Funder JW; Carey RM; Mantero F; et al The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2016.PMID 26934393
- [10]Kapur G; Baracco R Evaluation of hypertension in children. Curr Hypertens Rep, 2013.PMID 23904150
- [11]New MI; Geller DS; Fallo F; et al Monogenic low renin hypertension. Trends Endocrinol Metab, 2005.PMID 15808805
- [12]Garovic VD; Hilliard AA; Turner ST Monogenic forms of low-renin hypertension. Nat Clin Pract Nephrol, 2006.PMID 17066054