Paeds · endocrinology-diabetes-and-growth
Precocious puberty
Also known as Central precocious puberty · GnRH-dependent precocious puberty · Peripheral precocious puberty · GnRH-independent precocious puberty · Premature sexual development
A fellowship approach to precocious puberty: recognise early sexual development as the onset of secondary sexual characteristics before 8 years in girls and before 9 years in boys, split it immediately into central (GnRH-dependent) and peripheral (GnRH-independent) forms plus the normal variants, confirm with bone age and a basal-then-stimulated gonadotropin work-up, and match the treatment to the driver — GnRH analog for central disease, cause-directed therapy for peripheral causes — because accelerated bone maturation and a missed central nervous system lesion are the two preventable harms.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who refuses to treat the label "early puberty" and instead asks who is driving the axis. The child in front of you has breast budding or pubic hair and a growth spurt that worries the family, but the answer that decides every investigation and every drug sits upstream — in the hypothalamus and pituitary. If the axis is genuinely switched on, gonadotropins rise and a GnRH analog will switch it back off. If the axis is being bypassed by an autonomous ovary, testis, adrenal gland or exogenous steroid, gonadotropins are suppressed and a GnRH analog is useless. Holding that single distinction through the history, the examination, the laboratory panel and the counselling is what the examination rewards, because it is what protects the child's height and catches the CNS tumour. [1] [2]
Overview & Definition
Picture the parents who bring a six-year-old girl because they have noticed breast buds and a smell of body odour, or the four-year-old boy whose testicles have visibly grown. Each is asking the same question in a different body: has puberty started, and if it has, is it the real, central, gonadotropin-driven process or something mimicking it? Precocious puberty is the appearance of secondary sexual characteristics earlier than the accepted normal window — before 8 years in girls and before 9 years in boys — and it matters for two reasons that recur throughout the topic. First, unchecked sex steroids accelerate bone maturation and fuse the growth plates early, so the tall child becomes the short adult. Second, the early signal can be the first and only clue to a central nervous system lesion or an adrenal or gonadal source of autonomous hormone. [1] [2]
The definition carries a refinement that examiners probe deliberately. The traditional thresholds were chosen for their sensitivity, so they flag many girls whose course is benign and slowly progressive, particularly girls with higher body mass index and those of some ethnic backgrounds in whom thelarche naturally begins earlier. A practical refinement lowers the trigger for full evaluation: any girl with thelarche before 7 years (and some guidelines before 6 years) and any boy with any pubertal change at any age. The thresholds are a screening net, not a cliff edge — what matters is the tempo, the consonance of the sequence, and whether the bone age is running ahead of the child. [1] [2] [7]
Classification
The classification that changes management is mechanistic, not cosmetic: you are asking whether the hypothalamic–pituitary–gonadal axis is the driver. When the axis is genuinely switched on, the process is central (GnRH-dependent) and gonadotropins rise — the same biology as normal puberty, simply switched on too early. When a sex-steroid source operates outside the axis, the process is peripheral (GnRH-independent) and gonadotropins are suppressed by negative feedback, because the brain correctly senses that there is already too much hormone in the circulation. A third group, the early-puberty variants — premature thelarche, premature adrenarche and premature menarche — are not puberty at all but isolated, non-progressive features that usually need only observation. [1] [2]
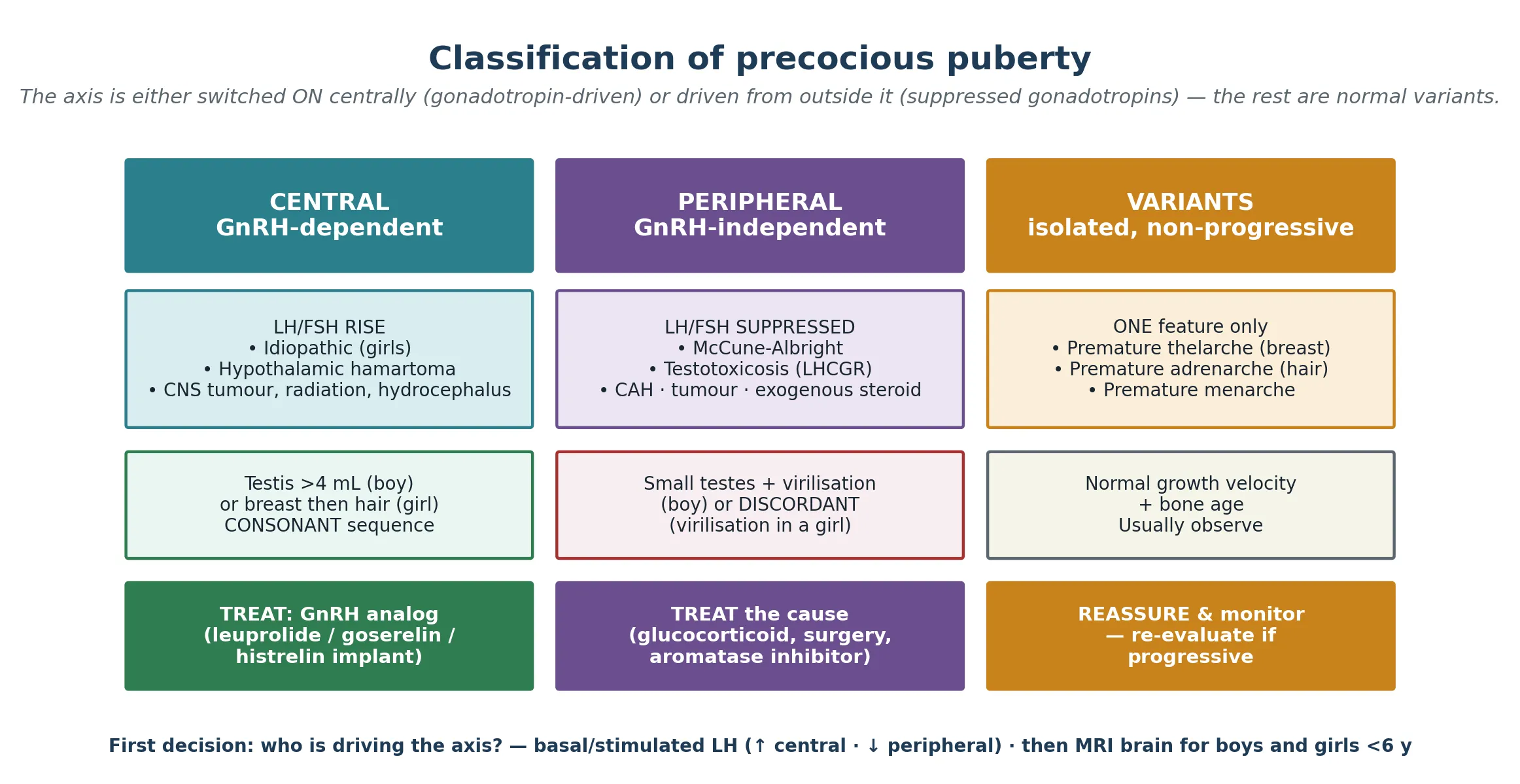
Central disease is itself divided by cause. In girls the large majority is idiopathic — the axis has simply been released from its childhood restraint too early, often in a child with higher body fat. In boys the balance inverts: a CNS lesion is found in roughly half or more, with a hypothalamic hamartoma the classic and most common, alongside tumours, the late effects of CNS radiation, malformations and hydrocephalus. Peripheral causes run by organ: the adrenal gland (congenital adrenal hyperplasia, adrenal tumour), the gonad (ovarian cyst or granulosa-cell tumour, Leydig or Sertoli cell tumour), a chorionic gonadotropin-secreting tumour, an activating receptor mutation (familial male-limited precocious puberty, McCune-Albright syndrome), or exogenous sex steroids. Keeping these columns separate is not academic: a GnRH analog works only for the central column. [1] [8] [9]
Epidemiology & Risk Factors
Central precocious puberty is roughly ten times more common in girls than in boys, and the reasons for that asymmetry are themselves the highest-yield epidemiology for the exam. In girls the process is usually idiopathic, and the secular trend toward earlier thelarche tracks closely with the rise in childhood obesity. Greater adiposity raises circulating leptin, which is permissive for the reactivation of hypothalamic kisspeptin and the GnRH pulse generator, so the heavier child crosses the pubertal threshold earlier. In boys the rarity of idiopathic disease is exactly what raises the pre-test probability of an organic CNS cause and is the reason every boy is imaged. [1] [6] [7]
The population shift toward earlier puberty is now well characterised. Longitudinal cohort data confirm that higher body mass index and rising adiposity through childhood associate with earlier puberty onset, particularly earlier thelarche in girls, and earlier work linked the secular trend in the United States to increasing weight and to ethnic variation. The clinical consequence is that a girl of seven or eight with isolated, slowly progressive breast development and a bone age close to her chronological age is now a common presentation — and is usually observed rather than treated. Exogenous exposure is the other recurring precipitant: topical or oral oestrogen, oral contraceptives, and the lavender and tea-tree oils reported to cause reversible prepubertal gynaecomastia and peripheral oestrogen effects. [6] [7] [4]
Pathophysiology
Normal puberty is a reactivation. After the minipuberty of infancy subsides, the hypothalamic GnRH pulse generator is held in check through childhood by a brake often called the gonadostat. At the usual age, kisspeptin neurons in the arcuate nucleus release that brake, pulsatile GnRH reaches the anterior pituitary, and the gonadotropins luteinising hormone and follicle-stimulating hormone drive the gonads to produce oestrogen in girls and testosterone in boys. The sex steroids then close the loop on growth plate maturation, the growth spurt, and the physical sequence of secondary sexual characteristics. Precocious puberty is what happens when that reactivation arrives early, or when the loop is short-circuited entirely. [1] [2]
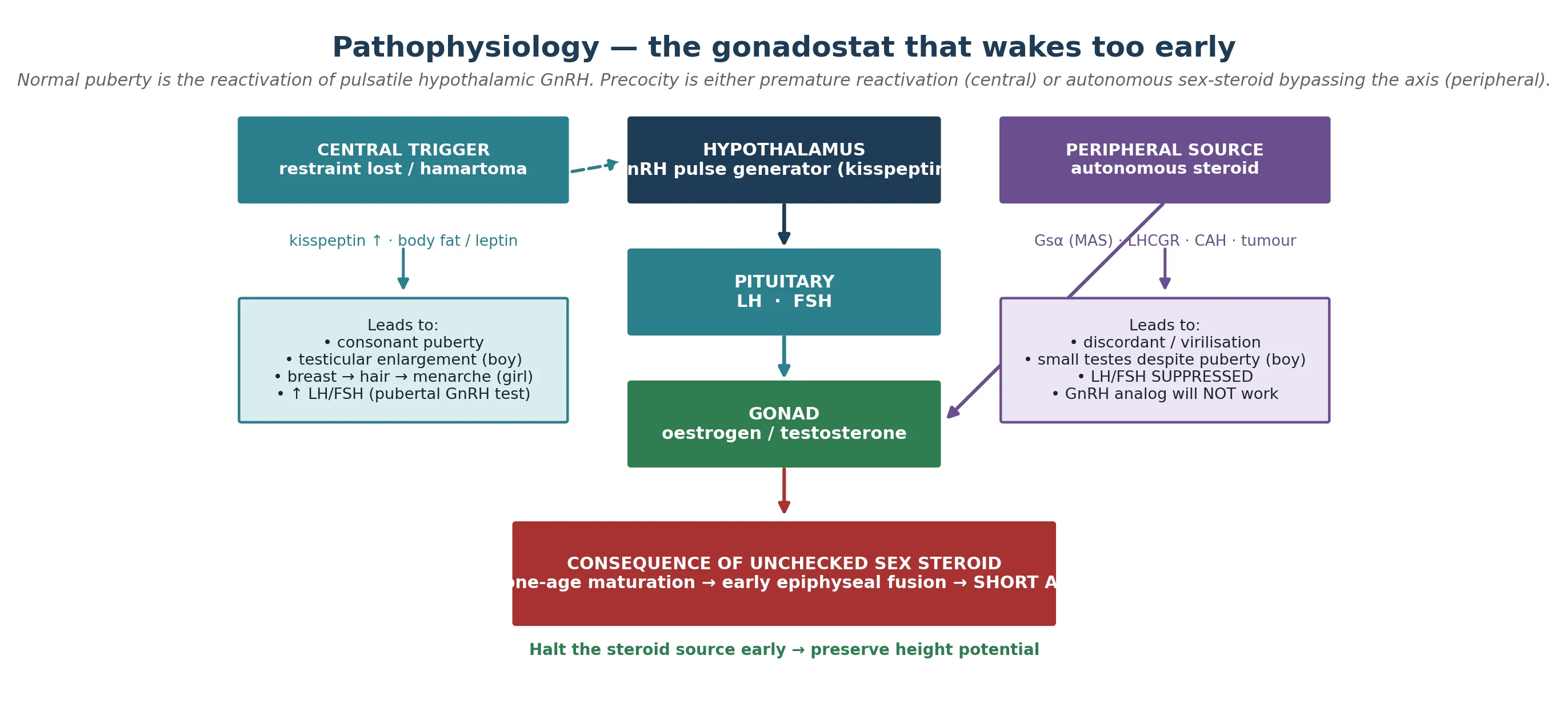
In central precocious puberty the restraint is lost early. In idiopathic girls the driver is the same permissive leptin-kisspeptin biology that governs normal puberty, simply triggered at a lower weight and age. A hypothalamic hamartoma is the archetypal organic cause: a benign congenital malformation near the tuber cinereum that either secretes GnRH directly or drives kisspeptin neurons, switching the axis on as if it were a second, ectopic pulse generator. In peripheral precocious puberty the axis is irrelevant. An activating, postzygotic Gs-alpha mutation in McCune-Albright syndrome keeps ovarian follicles producing oestrogen autonomously; an activating LHCGR mutation in familial male-limited precocious puberty (testotoxicosis) drives Leydig cells to make testosterone independent of luteinising hormone; and 21-hydroxylase deficiency in congenital adrenal hyperplasia shunts steroidogenesis into adrenal androgen excess. In every peripheral case the pituitary is suppressed, and that suppression is both the diagnostic signature and the reason a GnRH analog cannot work. [1] [8] [9]
Clinical Presentation
Read the presentation in two layers: which feature appeared first, and in what order the rest followed. In a girl with central disease thelarche (breast budding) usually comes first, followed by adrenarche (pubic and axillary hair), then the growth spurt, and finally menarche. That sequence is consonant — it runs in the normal pubertal order, just early — and its consonance is itself reassuring that the axis, not an outsider, is driving. In a boy with central disease the first sign is testicular enlargement beyond 4 mL or 2.5 cm in length, and that single sign is the most useful bedside discriminator of a gonadotropin-driven process, because the testis cannot grow without gonadotropins. [1] [2]
Peripheral disease announces itself differently. A girl may present with virilisation — clitoromegaly, a deepening voice, rapid muscle bulk, severe acne — which is discordant, an androgen pattern in the wrong body, and which points to a peripheral source such as congenital adrenal hyperplasia or an adrenal or gonadal androgen-secreting tumour. A boy may present with a virilised body but small, prepubertal testes, because the androgens are coming from the adrenal gland or a tumour rather than from gonadotropin-driven testicular growth. The exception is testotoxicosis, where the Leydig cells enlarge the testes themselves but out of proportion and out of sequence. Associated features in either form include accelerated linear growth, adult body odour, mood and behavioural change, acne, and, in girls, vaginal bleeding — which can be distressing when menarche arrives in a six-year-old. [1] [8] [9]
Differential Diagnosis
The differential is best built around the presenting feature, because the first feature tells you which hormone is in excess. Early breast development raises central precocious puberty, the isolated and usually non-progressive variant of premature thelarche, an oestrogen-secreting ovarian cyst or granulosa-cell tumour, exogenous oestrogen exposure, and McCune-Albright syndrome. Early pubic or axillary hair raises premature adrenarche, congenital adrenal hyperplasia, and an adrenal or gonadal androgen-secreting tumour. The job of the clinician is to separate the variant (isolated, slow, normal bone age) from true precocity (progressive, with bone-age advance) and then to split true precocity into its central and peripheral drivers. [1] [2] [5]
McCune-Albright syndrome earns its own place in the differential because it is the classic peripheral cause and the one examiners reach for. The postzygotic activating Gs-alpha mutation produces the triad of polyostotic fibrous dysplasia, large irregular cafe-au-lait macules with the "coast of Maine" border, and autonomous endocrine hyperfunction — most often peripheral precocious puberty in girls. The vaginal bleeding that brings the child to attention can be dramatic and early, and the bone pain or deformity and the skin pigmentation are the clues that the ovary is not the whole story. Congenital adrenal hyperplasia sits on the differential of any virilised child: the salt-wasting classic form declares itself in the newborn with hyponatraemia, hyperkalaemia and vomiting, while the simple-virilising and non-classic forms present later with early androgen signs and are unmasked by a 17-hydroxyprogesterone level. [8] [9] [5]
Clinical & Bedside Assessment
The bedside assessment begins with the growth chart, because growth velocity is the most objective measure of how hard the sex steroids are pulling. Plot height, weight and body mass index, record both parents' heights and calculate the mid-parental target height, and measure growth velocity over at least four to six months so the trajectory is real rather than a single point. A child whose height has jumped two centile bands in a year is being driven by sex steroids, and the magnitude of the jump is a measure of how urgently the bone age is advancing. [1] [2]
The pubertal examination is performed and recorded formally, not in passing. In girls, stage the breasts (Tanner B1 to B5) and the pubic hair (Tanner P1 to P5) separately, because dissociation between them is informative. In boys, stage the genitals and, crucially, measure testicular volume with a Prader orchidometer — a volume above 4 mL or a length above 2.5 cm is the threshold of central activation. Examine for the red flags of an organic driver: neurological signs and visual fields for a CNS lesion, cafe-au-lait macules and bony deformity for McCune-Albright, an abdominal mass for an adrenal or ovarian tumour, an asymmetric or palpable testis for a gonadal tumour, and signs of androgen excess against oestrogen excess to judge consonance. Take a careful exposure history — creams, supplements, essential oils, parental medications. [1] [2] [4]
Investigations
The single most informative first test is the bone age — a radiograph of the left hand and wrist — because it quantifies the biological cost of the sex-steroid exposure. A bone age advanced by more than two years above the chronological age signals true precocity that is eroding height potential and usually warrants treatment; a bone age close to the chronological age supports a variant or a slowly progressive course that can be watched. The bone age does not, however, tell you who is driving the axis, and that question is settled by the gonadotropins. [1] [2]
The baseline endocrine panel begins the central-versus-peripheral split at the bedside. Measure luteinising hormone, follicle-stimulating hormone, oestradiol (in girls) or testosterone (in boys), dehydroepiandrosterone sulphate, androstenedione, 17-hydroxyprogesterone and thyroid function. A clearly pubertal luteinising hormone already points to central disease; a suppressed luteinising hormone and follicle-stimulating hormone with a high sex steroid points to peripheral disease. When the basal gonadotropins are equivocal, the GnRH stimulation test — or a GnRH-agonist stimulation using leuprolide — is the gold standard for central precocious puberty: a pubertal luteinising hormone response, typically a peak above 4 to 5 IU per litre, confirms that the axis is genuinely switched on. A pelvic ultrasound assesses uterine size and ovarian morphology in girls, and a testicular ultrasound is mandatory for any asymmetric or palpable testis. [1] [2] [3]
When is a brain MRI mandatory in precocious puberty?
Brain and pituitary MRI is indicated for every boy with central precocious puberty, for every girl under 6 years, and for any child with neurological signs, visual symptoms, or an atypical or rapidly progressive course. The hypothalamic hamartoma is the most common lesion found. Girls between 6 and 8 years with a clearly idiopathic, typical course are the one group in whom imaging is guided by clinical judgement rather than mandated. [1] [2]
Management — Resuscitation
Precocious puberty is rarely a same-minute emergency, but two of its causes are, and both announce themselves through the virilised infant. Salt-wasting congenital adrenal hyperplasia in a newborn or young infant presents with hyponatraemia, hyperkalaemia, hypoglycaemia and vomiting, and is stabilised with intravenous fluids, hydrocortisone and glucose before the endocrine work-up is complete. A high 17-hydroxyprogesterone confirms 21-hydroxylase deficiency, but treatment of the salt-wasting crisis begins on clinical and biochemical grounds without waiting for the confirmatory result. [9]
The other acute presentations are structural. A central nervous system tumour may declare itself with headache, visual loss or altered conscious state, and demands urgent imaging with neurosurgical or oncological input. Heavy or prolonged vaginal bleeding — from a large oestrogen-secreting ovarian cyst or from McCune-Albright — can be acutely distressing in a small child and occasionally requires acute hormonal or surgical control. Across all of these, there is a psychosocial urgency that belongs to the first visit: the child who looks years older than their peers is frightened and visible, and early psychological support is part of stabilisation, not an afterthought to the work-up. [1] [8]
Management — Definitive & Stepwise
Definitive management is built on the framework of confirm, classify, image, and match the treatment to the driver. For central progressive precocious puberty, the treatment is a GnRH analog — leuprolide acetate given as an intramuscular depot monthly or three-monthly, goserelin as a subcutaneous implant, or triptorelin — or a histrelin subcutaneous implant that releases drug for up to a year. These agents down-regulate pituitary GnRH receptors, suppress gonadotropins, halt pubertal progression, slow bone-age acceleration and preserve adult height. A modern phase 3 study of a six-monthly leuprolide depot confirms sustained suppression and an acceptable long-term safety profile. The indication is progressive disease with compromise of predicted adult height or significant psychosocial impact; not every child needs treatment, and slowly progressive disease is often observed. [3] [10]
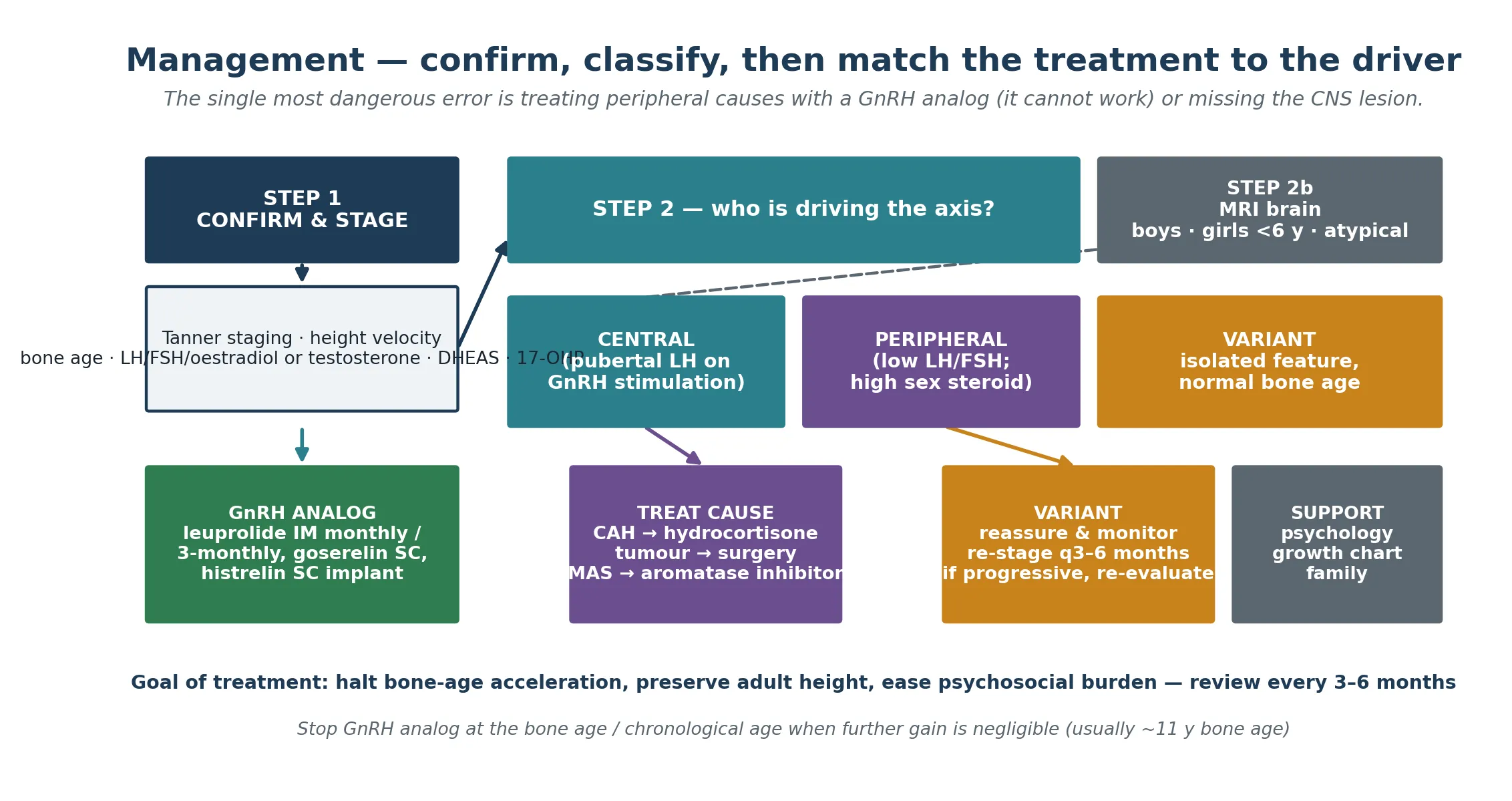
For peripheral disease, the treatment is at the source, and a GnRH analog is explicitly not the answer. Congenital adrenal hyperplasia is treated with glucocorticoid replacement — hydrocortisone in children — to suppress adrenocorticotropic hormone and the consequent adrenal androgen excess, with mineralocorticoid (fludrocortisone) and salt supplementation in the salt-wasting forms. An androgen- or oestrogen-secreting tumour is treated surgically. McCune-Albright peripheral puberty is treated with an aromatase inhibitor such as letrozole or anastrozole, or with tamoxifen, under specialist guidance. Testotoxicosis is managed with a combination of an antiandrogen (spironolactone or bicalutamide) and an aromatase inhibitor, because the autonomous testis cannot be switched off at the pituitary. The variants — premature thelarche, premature adrenarche, premature menarche — are reassured and monitored with re-staging every three to six months, and re-evaluated if they become progressive or if growth or bone age accelerate. [9] [8] [3]
Treatment by driver — specialist-initiated, weight-based, confirm current dosing
Monitoring is shared between the paediatric endocrinologist and the general paediatrician or family doctor. At each visit, every three to six months, measure growth velocity and re-stage puberty, and reassess bone age every six to twelve months to confirm that maturation has slowed. The GnRH analog is continued until the bone age and chronological age reach the point where further height gain is negligible — commonly around a bone age of 11 years in girls — at which point treatment is stopped and puberty resumes, now in step with peers. Counselling the family through this stopping point is as important as the starting decision. [3] [10]
Specific Subtypes & Scenarios
Idiopathic central precocious puberty in a girl is the commonest scenario and the prototype of central disease. The history is thelarche before 7 or 8 years with a consonant sequence and a growth spurt, the bone age is advanced, and the GnRH stimulation test is pubertal. When the course is progressive and the predicted adult height is compromised, a GnRH analog preserves height and eases the psychosocial burden; when it is slowly progressive with a bone age close to chronological age, observation is reasonable. [1] [2]
Central disease from a hypothalamic hamartoma combines peripheral puberty (which is in fact central, driven by the ectopic GnRH or kisspeptin tissue) with gelastic (laughing) seizures in some children. The puberty is managed with a GnRH analog; the hamartoma itself, when it causes intractable seizures, may warrant specialist neurosurgical or radiosurgical disconnection. McCune-Albright syndrome delivers peripheral puberty through autonomous ovarian oestrogen, set against polyostotic fibrous dysplasia and the coast-of-Maine cafe-au-lait macules; management is an aromatase inhibitor or tamoxifen plus a specialist multidisciplinary team for the bone and other endocrine disease. [1] [8]
Testotoxicosis (familial male-limited precocious puberty) is the activating LHCGR mutation passed father-to-son: a boy with virilisation, enlarged testes out of sequence, high testosterone, and suppressed gonadotropins. Because the Leydig cell is autonomous, treatment combines an antiandrogen with an aromatase inhibitor rather than a GnRH analog. Congenital adrenal hyperplasia — whether salt-wasting, simple-virilising or non-classic — drives peripheral androgen excess through 21-hydroxylase deficiency and is managed with glucocorticoid (and mineralocorticoid where indicated) to suppress the adrenal axis, following the Endocrine Society guideline. The variants close the section: premature thelarche, premature adrenarche and premature menarche are isolated and non-progressive, and are reassured and monitored rather than treated. [9] [8] [5]
Complications & Pitfalls
The cardinal complication is short adult stature. Unchecked sex steroids — central or peripheral — accelerate bone-age maturation and fuse the growth plates early, so the child who is tall for their age becomes the adult who is short for their family. Timely GnRH-analog treatment of progressive central disease recovers most of the lost height potential, which is the principal measurable justification for treating a child whose only "symptom" is early development. The matching cardinal error is missing the CNS lesion, concentrated in boys and in girls under 6 years, all of whom warrant imaging. [1] [10]
The therapeutic errors are the mirror image of the classification. Treating a peripheral cause with a GnRH analog is futile — the suppressed axis cannot be suppressed further — and it surrenders the height and the chance to treat the driver while creating the illusion of action. Treating slowly progressive or variant disease with depot injections over-treats a child who would have done well without them, at real cost and with the psychosocial labelling of a medicalised childhood. The procedural pitfalls of GnRH-analog therapy are injection-site reactions and sterile abscess at the depot, the transient hormone flare at initiation, and the long-term monitoring burden. The recurring system failure is simpler: the family lost to follow-up in whom bone-age acceleration goes unrecognised between visits. [3] [2]
Prognosis & Disposition
Prognosis is set by three things: the driver, the age and tempo at onset, and the timeliness of treatment. Idiopathic central disease in a girl carries the best prognosis — GnRH-analog treatment started early in a progressive course recovers most of the lost height potential and allows puberty to resume in step with peers. Organic central disease and the peripheral causes depend on the underlying lesion or syndrome: a resectable tumour, well-controlled congenital adrenal hyperplasia and treated McCune-Albright each carry their own trajectory, but all demand lifelong specialist surveillance for the height, the bones and the other endocrine axes. [1] [8] [9]
Disposition is shared care. The general paediatrician or family doctor holds the growth chart and the psychosocial context, the paediatric endocrinologist holds the diagnosis and the drug, the psychologist supports the child who looks older than their peers, and — for organic causes — the neurosurgeon or oncologist holds the lesion. Transition to adult endocrinology matters mainly for the syndromic and peripheral causes (McCune-Albright, congenital adrenal hyperplasia) rather than for treated idiopathic central disease. The honest message to the family is favourable: in treated central disease, adult height and fertility are preserved, and the early-development gap closes as the child grows into their body. [2] [3]
Special Populations
The same condition behaves differently across populations because recognition and access are unevenly distributed. In remote and Indigenous communities, and in migrant and refugee families, the interval between the first pubertal change and a measured gonadotropin panel is longer, the brain MRI is harder to access, and the depot injections that hold the height are harder to sustain at the required frequency. The practical response is an outreach-supported, telehealth-augmented endocrine service, a written and location-specific plan, and a low threshold to measure the panel in any child with early development rather than waiting for the referral to mature. [1] [2]
The child with a co-occurring neurodevelopmental disability carries a magnified psychosocial burden, because the mismatch between a maturing body and a younger mind is sharper and the capacity to process it is less; educational and psychological support are part of the medical plan, not adjacent to it. The boy with central precocious puberty is a special population in his own right, with a roughly fifty-percent-or-greater yield of CNS pathology that mandates MRI and distinguishes him from the idiopathic majority of girls. The adolescent who was treated in childhood reintegrates with peers once treatment stops and puberty resumes, and counselling ahead of that transition prevents the risk-taking behaviours that can accompany an early-maturing body in a still-maturing mind. Care delivered here should never frame early development as a deficit; it should support the child's identity, autonomy and dignity through a frightening and highly visible condition. [1] [2]
In Australia and New Zealand, central precocious puberty is managed through the state paediatric endocrine services, with GnRH-analog therapy initiated by a paediatric endocrinologist and shared monitoring with the general paediatrician or GP. Access to depot therapy and to timely brain MRI is uneven across rural and remote regions, which is the principal equity issue. Telehealth-endocrinology outreach and aeromedical retrieval services narrow but do not close the gap. [1] [3]
Evidence, Guidelines & Regional Differences
The evidence base rests on four pillars. The international consensus on GnRH-analog use in children (Bangalore Krishna 2019) sets out the indications, the agents and routes, and the monitoring of efficacy and safety, and is the structural foundation of modern treatment. The contemporary reviews (Carel and Leger 2008; Kaplowitz 2020) define the central-versus-peripheral distinction, the lower-yield evaluation of girls aged 6 to 8 years, and the decision to treat or observe. The cause-specific guidelines — the Endocrine Society congenital adrenal hyperplasia guideline (Speiser 2018) and the McCune-Albright review (Dumitrescu and Collins 2008) — govern the management of the peripheral causes. The epidemiology and treatment-outcome literature explains the secular shift and confirms the height benefit of depot therapy. [3] [1] [2] [9]
The age thresholds for evaluation differ modestly across regions. The Endocrine Society and the BSPED (British Society for Paediatric Endocrinology and Diabetes) endorse a lower trigger for full evaluation in girls (thelarche before 6 to 7 years, depending on context) than the traditional "before 8 years", reflecting the secular trend and the poor sensitivity of the older threshold. All major guidelines agree that every boy with precocious puberty warrants evaluation and brain imaging. The GnRH-analog agents and routes vary by national availability — leuprolide depots, goserelin, triptorelin and the histrelin implant — but the principle of matching the treatment to the driver is universal. [1] [2] [3]
The leuprolide depot trial (Klein 2026) adds recent long-term safety and efficacy data for the three- and six-monthly depots, reinforcing that less-frequent dosing does not sacrifice suppression. The obesity-and-puberty cohort (Li 2022) and the earlier secular-trend work (Kaplowitz and Herman-Giddens 2001) explain the population shift toward earlier thelarche in girls. The controversies that remain are familiar: how far to lower the evaluation threshold for girls without over-diagnosing slowly progressive disease; the long-term bone-density and reproductive effects of GnRH-analog treatment; and the cost and access inequity of depot therapy that concentrates the height benefit among those who can reach the service. [10] [6] [7]
Exam Pearls
The thresholds and the clinical refinement are the highest-yield facts: before 8 years in girls and before 9 years in boys are the screening thresholds, but evaluate any girl with thelarche before 7 years and any boy with pubertal change at any age. The central-versus-peripheral split is settled by the gonadotropins — a pubertal luteinising hormone response on GnRH stimulation confirms central disease, while a suppressed luteinising hormone and follicle-stimulating hormone with a high sex steroid confirms peripheral disease. The brain MRI is mandatory for every boy and for every girl under 6 years, and the hypothalamic hamartoma is the classic lesion found. [1] [2]
PERIPHERAL causes — the GnRH-independent list
A GnRH analog works only for central disease, and that single rule is the most testable point in the management of precocious puberty. Peripheral causes must be treated at their source — glucocorticoid for congenital adrenal hyperplasia, surgery for a tumour, an aromatase inhibitor for McCune-Albright, an antiandrogen plus an aromatase inhibitor for testotoxicosis. And the mechanism of McCune-Albright — a postzygotic activating Gs-alpha mutation, with the triad of polyostotic fibrous dysplasia, coast-of-Maine cafe-au-lait macules and autonomous endocrine hyperfunction — is the classic peripheral cause that examiners reward a candidate for naming in full. [8] [9] [3]
References
- [1]Carel JC, Léger J. Clinical practice. Precocious puberty. N Engl J Med, 2008.PMID 18509122
- [2]Kaplowitz PB. Update on Precocious Puberty: Who Should Be Treated? Adv Pediatr, 2020.PMID 32591066
- [3]Bangalore Krishna K, Fuqua JS, Rogol AD, Klein KO, et al. Use of Gonadotropin-Releasing Hormone Analogs in Children: Update by an International Consortium. Horm Res Paediatr, 2019.PMID 31319416
- [4]Henley DV, Lipson N, Korach KS, Bloch CA. Prepubertal gynecomastia linked to lavender and tea tree oils. N Engl J Med, 2007.PMID 17267908
- [5]Rosenfield RL. Normal and Premature Adrenarche. Endocr Rev, 2021.PMID 33788946
- [6]Li Y, Ma T, Ma Y, Gao D, et al. Adiposity Status, Trajectories, and Earlier Puberty Onset: Results From a Longitudinal Cohort Study. J Clin Endocrinol Metab, 2022.PMID 35779008
- [7]Kaplowitz PB, Slora EJ, Wasserman RC, Pedlow SE, Herman-Giddens ME. Earlier onset of puberty in girls: relation to increased body mass index and race. Pediatrics, 2001.PMID 11483799
- [8]Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis, 2008.PMID 18489744
- [9]Speiser PW, Arlt W, Auchus RJ, Baskin LS, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2018.PMID 30272171
- [10]Klein KO, Mauras N, Nayak S, Sunil B, Soliman AM, Kansra AR. Long-term Safety, Efficacy, and PROs: Phase 3 Study of Leuprolide Acetate 6-month IM Depot in Central Precocious Puberty. J Endocr Soc, 2026.PMID 41623908