Paeds · endocrinology-diabetes-and-growth
Rickets and metabolic bone disease
Also known as nutritional rickets · vitamin D deficiency rickets · hypocalcaemic rickets · X-linked hypophosphataemia · vitamin D-dependent rickets
A fellowship approach to rickets and metabolic bone disease in children: defective mineralisation of growing bone that is almost always preventable, the calcipenic-versus-phosphopenic classification that the biochemistry reveals in seconds, the vitamin D cascade and where it breaks, the symptomatic hypocalcaemic infant who needs intravenous calcium before vitamin D, and the lifelong pathway for hereditary forms such as X-linked hypophosphataemia.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Picture the child who walks in. A toddler is brought in because the legs have bowed since she started walking, the wrists look thick, and the fontanelle is still open at eighteen months. Or a six-month-old, exclusively breastfed by a veiled mother in a southern winter, is brought to the emergency department fitting, with a calcium of 1.4 and a cholecalciferol level that is unmeasurable. Or a school-age child with short stature and repeated dental abscesses turns out to have X-linked hypophosphataemia that was never nutritional at all. The fellowship task in each is to read the calcium, phosphate, and parathyroid hormone before the vitamin D level, and to treat the cause rather than reflexively loading vitamin D. [8] [9]
C-A-P-P for the rickets work-up
Overview & Definition
Rickets is the failure of mineralisation at the growing cartilage of the epiphyseal plate, and the companion lesion in bone already formed is osteomalacia, the failure of mineralisation of osteoid. In a growing child the two coexist, and the clinical and radiographic picture reflects both: the softened growth plate widens and bows under load, while the unmineralised osteoid accumulates and deforms. The unifying problem is a mineral shortfall at the mineralisation front, and that shortfall is driven by a deficiency of calcium, of phosphate, or of the vitamin D that delivers calcium to the gut. [5] [7]
The historical weight of the disease is large. Before supplementation and fortification, nutritional rickets was among the commonest causes of disability in children, and its disappearance from high-income populations in the twentieth century was one of public health's clearest victories. Its return in the twenty-first century, in exclusively breastfed infants, in dark-skinned and veiled populations at high latitude, and in refugee and migrant families, is documented across Europe, North America, and Australasia, and it is the reason every general paediatrician must still own the topic. [8] [6]
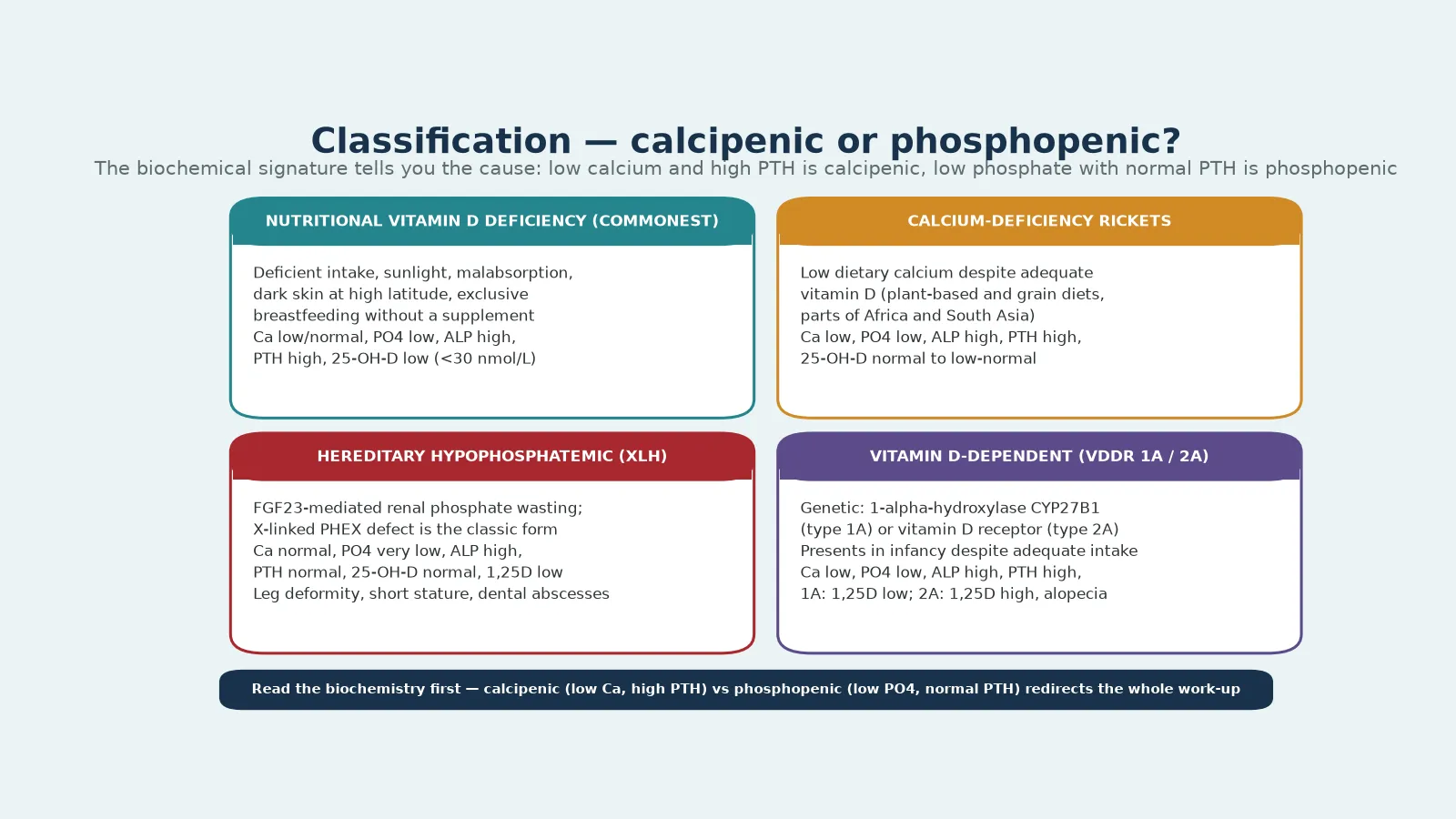
What makes rickets a fellowship-level subject rather than a single-disease memory is that the mineralisation failure has many causes that look identical on a radiograph and nearly identical on examination, yet demand completely different treatment. Nutritional vitamin D deficiency is the commonest and the most preventable, but calcium deficiency, genetic defects of vitamin D metabolism (vitamin D-dependent rickets), and renal phosphate wasting (X-linked hypophosphataemia) all produce the same widened, frayed metaphysis. Distinguishing them is a biochemical exercise, and the biochemistry is read in seconds once you know the cascade. [5] [1]
Classification
The classification that earns marks separates rickets by the mineral that is short at the growth plate. Calcipenic rickets arises when too little calcium reaches the mineralisation front, and it carries a low or low-normal calcium, a high alkaline phosphatase, and a raised parathyroid hormone as the gland works to compensate. Phosphopenic rickets arises when phosphate is the limiting substrate, and it carries a low phosphate with a normal or low parathyroid hormone, because the parathyroid axis is not the problem. Reading that single contrast — is the calcium low or is the phosphate low — orients the whole differential before the vitamin D level returns. [5] [1]
Within calcipenic rickets, nutritional vitamin D deficiency is overwhelmingly the commonest cause. It reflects deficient intake, deficient sunlight exposure, malabsorption, or the increased demand of rapid growth, and its biochemical signature is a low 25-hydroxyvitamin D with a low or low-normal calcium, a low phosphate, a high alkaline phosphatase, and a high parathyroid hormone. Calcium-deficiency rickets sits beside it: dietary calcium is the limiter even when vitamin D is adequate, classically in children on low-calcium, high-phytate, plant-based diets in parts of Africa and South Asia, and it produces the same calcipenic signature with a vitamin D level that is normal or only mildly low. [11] [7]
The genetic calcipenic forms declare themselves in infancy despite adequate intake and supplementation. Vitamin D-dependent rickets type 1A is a defect of the renal 1-alpha-hydroxylase (CYP27B1) that cannot make 1,25-dihydroxyvitamin D, so the active hormone is low while the precursor is normal or high. Type 2A is a defect of the vitamin D receptor, so the active hormone is high but the tissue cannot respond, and alopecia is a distinctive clue. These children need calcitriol, often in high doses, rather than standard cholecalciferol. [5] [12]
Phosphopenic rickets is dominated by X-linked hypophosphataemia, an FGF23-mediated renal phosphate wasting caused by PHEX mutation, and the most common inherited form of rickets. Its signature is a markedly low phosphate with a normal calcium, a normal or low parathyroid hormone, and a normal 25-hydroxyvitamin D, and the 1,25-dihydroxyvitamin D is inappropriately low because FGF23 suppresses the 1-alpha-hydroxylase. Other hereditary hypophosphataemias and tumour-induced osteomalacia share this FGF23-driven signature, and none of them heal with vitamin D alone. [9] [5]
Epidemiology & Risk Factors
Nutritional rickets is a disease of the interface between biology and environment, and its epidemiology tracks sunlight, skin, diet, and latitude. In high-income countries the resurgence is concentrated in exclusively breastfed infants, because human milk carries little vitamin D, and in infants of dark-skinned, veiled, or immigrant mothers, whose melanin reduces cutaneous vitamin D synthesis and whose cultural or climatic dress practices limit sun exposure. A winter birth in a temperate climate compounds all of these, and the typical case presents in late winter or early spring. [8] [3]
The Global Consensus on the prevention of nutritional rickets, published in 2016, reframed the public health target. It defined vitamin D sufficiency as a 25-hydroxyvitamin D above 50 nmol per litre, recommended a daily intake of 400 IU/day for infants 0–12 months and at least 600 IU/day from age 12 months through adolescence, and stated plainly that exclusive breastfeeding without supplementation is the single commonest identifiable cause of nutritional rickets where fortification is absent. The consensus also emphasised maternal vitamin D status, because a deficient mother delivers a deficient infant. [1] [2]
Globally the picture is broader. Calcium-deficiency rickets, where dietary calcium intake is the limiter, is well described in children with adequate sunlight and normal vitamin D status in parts of sub-Saharan Africa and South Asia, often on diets high in phytates that bind intestinal calcium. In these settings rickets coexists with normal 25-hydroxyvitamin D, and the treatment is calcium rather than vitamin D. This global variation is why a fellowship answer refuses to equate rickets with vitamin D deficiency. [11] [6]
The hereditary forms are individually rare but collectively important. X-linked hypophosphataemia has an incidence of about one in twenty thousand, and it is the most common genetic cause of rickets; because it is X-linked dominant, it affects both sexes, with variable severity. Vitamin D-dependent rickets types 1A and 2A are autosomal recessive and present in infancy. Premature infants form a further population, because they miss the third-trimestoral calcium and phosphate accretion and need supervised supplementation. [9] [4]
The numbers that anchor a fellowship answer
Pathophysiology
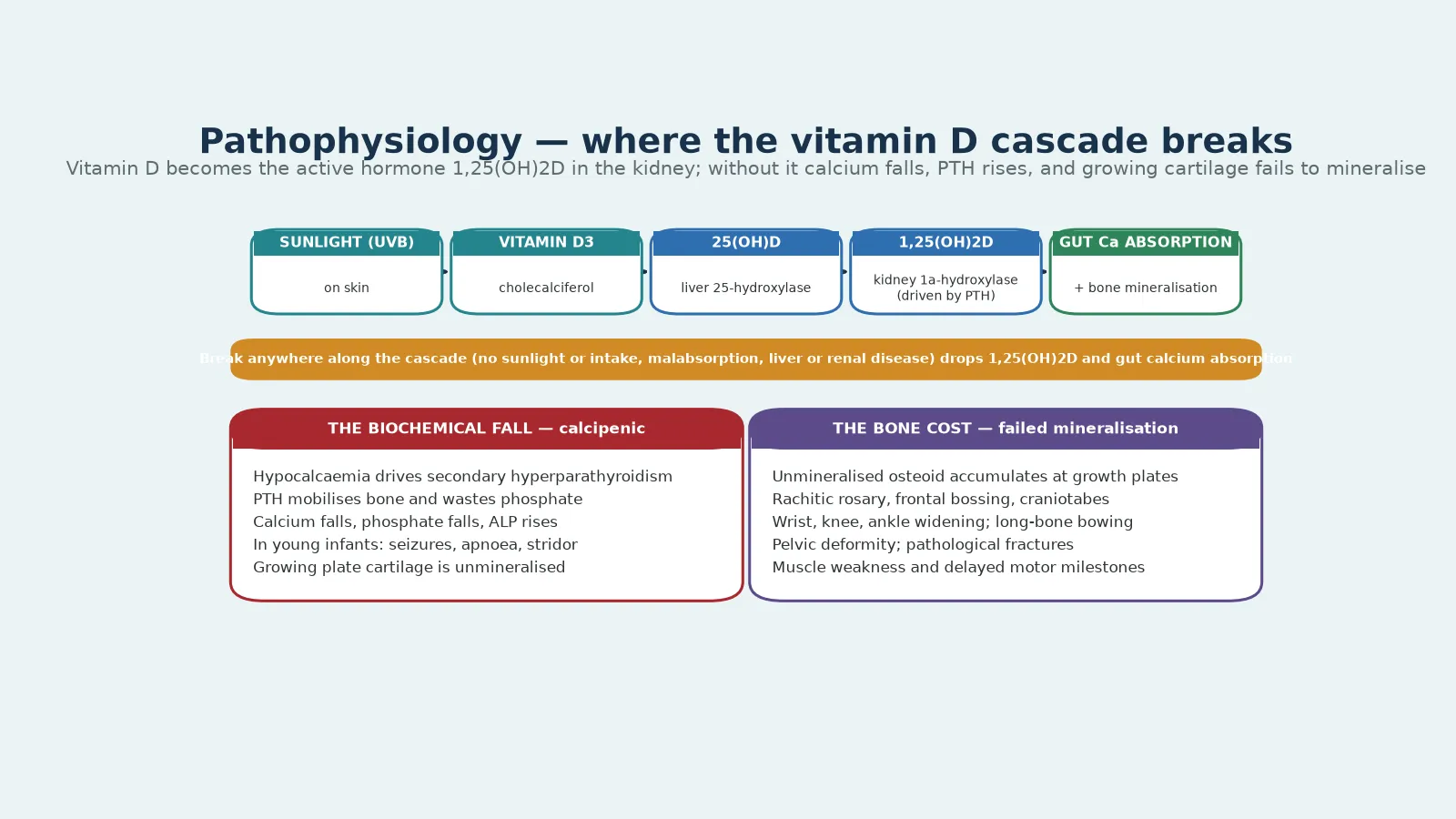
The pathophysiology turns on a single hormone cascade that delivers calcium to the mineralisation front, and a fellowship answer walks it from skin to bone. Ultraviolet B light acts on 7-dehydrocholesterol in the skin to form vitamin D3 (cholecalciferol), which the liver converts to 25-hydroxyvitamin D, the storage form measured in blood. The kidney then adds a second hydroxyl group through the 1-alpha-hydroxylase, regulated by parathyroid hormone, to produce 1,25-dihydroxyvitamin D, the active hormone. That active hormone acts on the gut to absorb calcium and phosphate, and it acts on bone to make mineral available for the growth plate. [2] [5]
When the cascade breaks anywhere along its length, calcium absorption falls, the serum calcium dips, and the parathyroid glands respond with secondary hyperparathyroidism. Parathyroid hormone mobilises calcium from bone and, critically, increases renal phosphate excretion, so phosphate falls in parallel. The product of serum calcium and phosphate drops below the threshold needed to mineralise osteoid and growth-plate cartilage, and the unmineralised matrix accumulates. The growth plate widens, the osteoid seams thicken, and the bone softens and bows under the load of walking and crawling. [5] [7]
The youngest infants show the consequence most dramatically. A rapid-growth infant with a deep vitamin D deficit can present with acute hypocalcaemia, because the parathyroid compensation has not yet caught up or has been overwhelmed. The clinical expression is seizure, apnoea, stridor, or tetany, and the biochemical expression is a low ionised calcium with a high parathyroid hormone and a high alkaline phosphatase. This is why the hypocalcaemic infant is treated as an emergency and why calcium is given before the vitamin D. [8] [1]
Phosphopenic rickets follows a different mechanism that does not pass through the parathyroid axis. In X-linked hypophosphataemia, loss of PHEX function raises FGF23, which acts on the kidney to suppress phosphate reabsorption and to suppress the 1-alpha-hydroxylase. Phosphate is wasted, the 1,25-dihydroxyvitamin D is inappropriately low, and the growth plate is starved of phosphate rather than calcium. The parathyroid hormone stays normal, there is no secondary hyperparathyroidism, and vitamin D loading alone cannot heal the bone. [9] [5]
Clinical Presentation
The presentation of nutritional rickets spans a spectrum from the asymptomatic biochemistry to the seizing infant, and the fellowship skill is to recognise both ends. The classic skeletal signs appear once mineralisation has failed for weeks to months: bowing of the legs (genu varum) or, less often, knock-knees (genu valgum) once the child is weight-bearing, swelling and widening at the wrists, knees, and costochondral junctions (the rachitic rosary), frontal bossing of the skull, delayed closure of the anterior fontanelle, craniotabes in the young infant, and dental delay with enamel defects. Proximal muscle weakness and hypotonia accompany the bone disease and contribute to the delayed motor milestones. [8] [5]
The infant who presents acutely is the must-not-miss end of the spectrum. A young, rapidly growing, exclusively breastfed infant of a vitamin D deficient mother may present between three and eighteen months with a seizure or a run of seizures, or with stridor from hypocalcaemic laryngospasm, or with apnoea, in the context of a normal or only mildly unwell intercurrent picture. The calcium is low, the phosphate is often low, and the alkaline phosphatase is markedly raised. The skeletal signs may be subtle or absent, and the seizure is the first manifestation; missing the biochemical cause sentences the child to recurrent seizures and, rarely, to cardiac compromise. [8] [1]
Hereditary rickets presents differently. X-linked hypophosphataemia declares itself once the child bears weight, with progressive lower-limb bowing, short stature that is disproportionate and leg-predominant, and a striking tendency to dental abscesses and periapical infection because the dentin is defective. Vitamin D-dependent rickets presents earlier, in infancy, often with hypocalcaemia and alopecia (in type 2A) despite adequate intake. The history of adequate supplementation and a normal or high vitamin D level, against a background of relentless rickets, is the clue that the cause is genetic rather than nutritional. [9] [5]
Differential Diagnosis
The differential splits into three questions: what else causes bowing and metaphyseal changes, what else causes a low calcium with a seizure, and what else looks like rickets on a radiograph. The first includes physiological bowing of toddlers, which is mild, symmetrical, and biochemical-normal, and Blount disease, an asymmetrical tibial growth-plate disorder that is not metabolic. These are separated from rickets by the biochemistry and the radiographic pattern: physiological bowing shows no metaphyseal fraying, and rickets does. [5] [8]
The biochemical differential matters because a normal vitamin D does not exclude rickets. Calcium-deficiency rickets produces the calcipenic signature with a normal 25-hydroxyvitamin D, and X-linked hypophosphataemia produces rickets with a normal calcium, normal parathyroid hormone, and normal vitamin D but a very low phosphate. Reading the calcium, phosphate, and parathyroid hormone before the vitamin D level prevents the error of dismissing a rachitic child because the vitamin D is normal. [11] [9]
A low alkaline phosphatase in a child who looks rachitic is the opposite trap and points to hypophosphatasia, an inherited deficiency of tissue-nonspecific alkaline phosphatase. Hypophosphatasia can present in infancy with rachitic features, premature loss of deciduous teeth, and fractures, but the alkaline phosphatase is inappropriately low rather than high, and the treatment is enzyme replacement with asfotase alfa, not vitamin D. Giving bisphosphonates, which are sometimes used in other bone diseases, is harmful in hypophosphatasia and worsens the mineralisation defect. [10] [5]
The remaining mimics include renal osteodystrophy from chronic kidney disease, which produces a mixed high and low parathyroid hormone picture depending on stage; metaphyseal chondrodysplasia, a primary bone dysplasia with normal biochemistry; osteogenesis imperfecta, where fractures rather than bowing dominate; and, when fractures are present in an infant, non-accidental injury, which is excluded by the biochemistry and radiographic pattern together with the clinical context. The discriminating move is always the paired biochemistry and radiograph. [5] [10]
Rickets and its mimics on biochemistry
Clinical & Bedside Assessment
The recognition move at the bedside is to lay hands on the growth plates. Examine the wrists, knees, ankles, and costochondral junctions for widening, palpate the skull for craniotabes and frontal bossing, look at the legs for bowing when the child stands, and assess the anterior fontanelle for delayed closure. Rachitic rosary is felt as a beaded, prominent ridge at the costochondral junctions, and the wrist swelling is often the first sign a parent has noticed. Look for muscle weakness and hypotonia, and assess the motor milestones, which are frequently delayed. [8] [5]
The history gathers the discriminators that point to a nutritional or a hereditary cause. Ask about feeding: exclusive breastfeeding without a supplement is the cardinal risk, and the introduction of unfortified solids prolongs it. Ask about sunlight, skin colour, clothing, latitude, and season, because dark skin, concealing clothing, high latitude, and winter all reduce cutaneous synthesis. Ask about the mother's vitamin D status, her dress, her diet, and her country of origin, because a deficient mother delivers a deficient infant. Ask about a family history of rickets, short stature, dental abscesses, or limb deformity, because X-linked hypophosphataemia is familial and sex-linked dominant. [3] [9]
The acutely presenting infant needs a different, faster assessment. The seizing or stridulous infant is examined for a patent airway, cardiorespiratory compromise, and signs of an alternative cause such as infection, and a blood gas, glucose, and electrolytes including ionised calcium are sent immediately. The calcium is low, the corrected calcium confirms hypocalcaemia, and the alkaline phosphatase is raised. The assessment is paired with the recognition that the family will need explanation, supplementation for siblings, and a coordinated public-health follow-up, because nutritional rickets in one infant often signals risk in the whole family. [8] [1]
The first visit also maps the practical context that determines whether treatment will succeed. Adherence to a daily supplement, access to a reliable supply of the correct preparation, transport for follow-up bloods and radiographs, interpreter needs, and health literacy all shape outcome as much as the prescription does. Identify a named coordinator early, plan the monitoring schedule, and arrange the public-health and primary-care links that will sustain the child and screen the family. [1] [3]
Investigations
The core panel is biochemical and is read as a pattern, not as individual numbers. Send a calcium (corrected, and ionised if acutely ill), phosphate, alkaline phosphatase, parathyroid hormone, and 25-hydroxyvitamin D, together with renal function and liver function to exclude chronic disease and malabsorption. The pattern is diagnostic: nutritional vitamin D deficiency shows a low 25-hydroxyvitamin D with a low or low-normal calcium, a low phosphate, a high alkaline phosphatase, and a high parathyroid hormone. Calcium-deficiency rickets shows the same calcipenic pattern with a normal 25-hydroxyvitamin D. [1] [5]
The measurement of 1,25-dihydroxyvitamin D is reserved for selected cases, because it is hard to interpret in isolation. It is low in vitamin D-dependent rickets type 1A (the 1-alpha-hydroxylase is absent) and inappropriately low in X-linked hypophosphataemia (FGF23 suppresses it), and it is high in vitamin D-dependent rickets type 2A (the receptor is defective and the hormone accumulates). A low phosphate with a normal parathyroid hormone and normal 25-hydroxyvitamin D should prompt measurement of renal phosphate handling, tubular reabsorption of phosphate, and, when XLH is likely, FGF23 and genetic testing for PHEX. [9] [5]
Radiographs confirm the diagnosis and stage its severity, and the wrist and knee are the standard views. The metaphyses are widened, splayed, frayed, and cupped, the growth plates are increased in depth, and in established disease the long bones show bowing and, sometimes, looser-zone (pseudofracture) lines. The radiograph also provides a baseline against which healing is judged, because the earliest sign of successful treatment is recalcification of the metaphyseal margin. [5] [6]
A practical point that earns marks: the alkaline phosphatase is the most useful single marker of activity and healing, and it should be checked at baseline and monitored through treatment. It falls within weeks of starting effective therapy, before the calcium and phosphate fully normalise and long before the radiograph heals. A persistently high alkaline phosphatase at three months means non-adherence, an unrecognised hereditary cause, or ongoing deficiency, and it prompts a review of the diagnosis rather than an endless dose escalation. [12] [1]
Management — Resuscitation
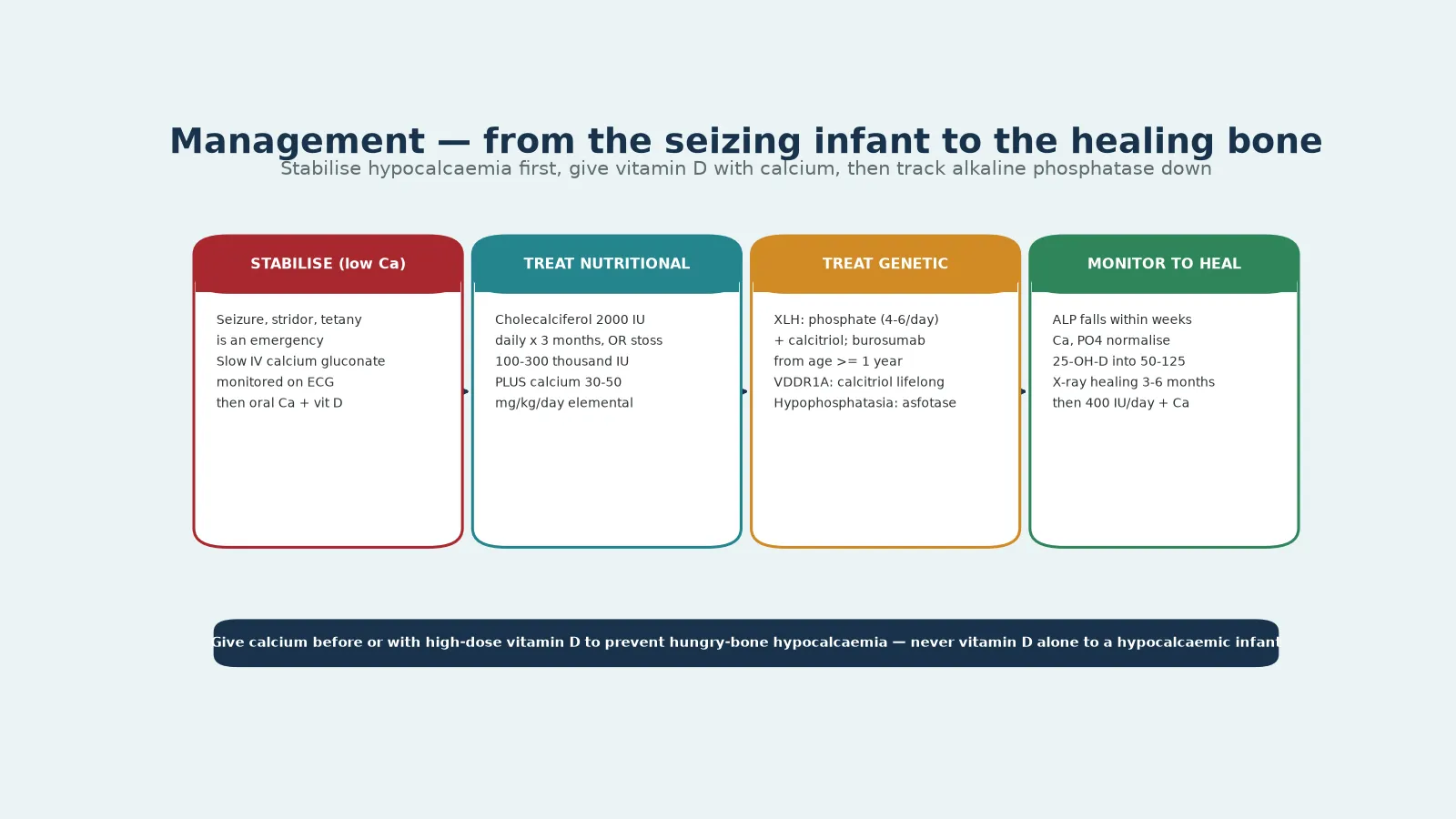
The resuscitation phase belongs to the symptomatic hypocalcaemic infant, and it is the scenario where the dose and the order of the drugs matter most. A seizing, stridulous, or tetanic infant with a low ionised calcium receives intravenous calcium gluconate, given slowly through a secure large-bore line with cardiac monitoring, because rapid infusion can cause bradycardia and arrest. The calcium is followed by oral calcium and cholecalciferol, and the family is counselled about the preventability of the disease and the need for treatment of siblings. [8] [1]
The principle that protects the hypocalcaemic infant is that calcium is given before or with the vitamin D, never vitamin D alone. High-dose vitamin D accelerates bone mineralisation and can precipitate or deepen hypocalcaemia through hungry bone, the rapid uptake of calcium and phosphate by the healing skeleton. Giving a stoss dose of vitamin D to a symptomatic hypocalcaemic infant without concurrent calcium is the classic avoidable error, and it can trigger seizures, arrhythmia, and, rarely, cardiac failure. The emergency sequence is calcium, then oral calcium with cholecalciferol, then monitoring. [1] [12]
Magnesium should be checked and corrected, because hypomagnesaemia produces functional hypoparathyroidism and renders the hypocalcaemia refractory until the magnesium is replaced. Acid-base status is reviewed, because alkalosis lowers the ionised calcium and worsens symptoms. The infant is admitted for observation, the seizures are terminated with calcium rather than with standard anticonvulsants (which are ineffective for hypocalcaemic seizures), and the transition to definitive therapy is planned once the calcium is stable. [5] [8]
Management — Definitive & Stepwise
Definitive treatment of nutritional rickets rests on cholecalciferol and calcium, and the Global Consensus gives two acceptable regimens. The first is a daily dose of cholecalciferol 2000 IU per day for three months, which is reliable and well tolerated. The second is a stoss regimen of a single oral dose, or a dose split over a day or two, of 100 000 to 300 000 IU, which is useful when adherence to a daily dose is uncertain. Both are given together with oral calcium at 30 to 50 mg per kilogram per day of elemental calcium, because giving vitamin D without calcium risks hungry-bone hypocalcaemia and slows healing. [1] [12]
Calcium-deficiency rickets is treated primarily with calcium, with vitamin D added if the 25-hydroxyvitamin D is also low. The response is brisk once the dietary calcium is corrected, and counselling about a calcium-adequate diet is part of the definitive plan, because the deficiency will recur if the diet reverts. In communities where dairy is scarce or unaffordable, the team works with a dietitian to find sustainable calcium sources, and the public-health dimension of the treatment is as important as the prescription. [11] [6]
Hereditary rickets follows separate pathways. X-linked hypophosphataemia is treated with oral phosphate in four to six divided daily doses together with calcitriol, the active hormone that bypasses the FGF23-suppressed 1-alpha-hydroxylase; burosumab, a monoclonal antibody against FGF23, is now licensed from age one year and offers an alternative that normalises phosphate without the renal and parathyroid complications of conventional phosphate. Vitamin D-dependent rickets type 1A is treated with lifelong calcitriol, because the 1-alpha-hydroxylase is absent; type 2A is harder, needing high-dose calcitriol or calcium infusion in refractory cases. [9] [5]
The treatment and monitoring sequence for nutritional rickets
Confirm the cause: calcipenic biochemistry with low 25-hydroxyvitamin D supports nutritional vitamin D deficiency
Start cholecalciferol 2000 IU daily for 3 months (or stoss 100 000–300 000 IU) PLUS oral calcium 30–50 mg/kg/day elemental
Check alkaline phosphatase and calcium at 4–6 weeks: the alkaline phosphatase should be falling
Repeat the wrist or knee radiograph at 3 months: look for recalcification of the metaphyseal margin
Confirm healing by 6 months, then move to maintenance 400 IU/day plus a calcium-adequate diet
Specific Subtypes & Scenarios
X-linked hypophosphataemia deserves its own pathway because it is the most common hereditary rickets and because its management is specialised. The diagnosis rests on a persistently low phosphate with renal phosphate wasting, a normal parathyroid hormone, and a normal 25-hydroxyvitamin D, confirmed by a PHEX mutation or a characteristic family history. The child is short, with disproportionate leg bowing and a characteristic tendency to dental abscesses, and the management combines endocrinology, nephrology, orthopaedics, and dentistry over a lifetime. Burosumab has changed the landscape by neutralising FGF23, normalising phosphate, and improving growth and rickets scores in children from age one year. [9] [5]
Vitamin D-dependent rickets is rarer but instructive. Type 1A, from CYP27B1 mutation, presents in infancy with hypocalcaemia and rickets despite adequate intake, a normal or raised 25-hydroxyvitamin D, and a low 1,25-dihydroxyvitamin D; it responds to calcitriol. Type 2A, from vitamin D receptor mutation, presents similarly but with a high 1,25-dihydroxyvitamin D and often alopecia, and it is harder to treat because the receptor itself is defective. The lesson is that a rachitic infant with a normal or high vitamin D level and a clear supplement history is not failing to adhere; the cause is genetic and the treatment is different. [5] [12]
The preterm infant is a distinct scenario. The bulk of calcium and phosphate accretion occurs in the third trimester, so a very preterm infant is born mineral-deficient and at risk of osteopenia of prematurity, a metabolic bone disease that overlaps with rickets. Supervised supplementation of calcium, phosphate, and vitamin D, careful attention to total parenteral nutrition mineral content, and monitoring of alkaline phosphatase and phosphate are the pillars, and the AAP guidance on enterally fed preterm infants specifies the targets. Fractures on routine handling in a preterm infant are the late-warning sign. [4] [5]
Hypophosphatasia is the subtype that is most easily mistaken for rickets and most easily harmed by the wrong treatment. The adult and childhood forms present with rickets-like features, premature loss of deciduous teeth, and fractures, but the alkaline phosphatase is low and the vitamin D and parathyroid hormone are normal. Enzyme replacement with asfotase alfa has transformed the outlook for the perinatal and infantile forms, and the vital safety message is that bisphosphonates, which might be considered for unexplained fractures, worsen the mineralisation defect in hypophosphatasia and must be avoided. [10] [5]
Complications & Pitfalls
The complications divide into those of the disease and those of its treatment. The disease itself, untreated or late-treated, produces progressive limb deformity, short stature, dental disease, pathological fractures, and, in the acutely hypocalcaemic infant, seizures and, rarely, cardiac compromise. The deformity may require orthopaedic correction once the metabolic disease is controlled, because the bone must be biochemically healed before any osteotomy is attempted; operating on metabolically active rickets risks recurrence and non-union. [5] [9]
The treatment complications are preventable and are the ones a fellowship answer names. Hungry-bone hypocalcaemia follows high-dose vitamin D given without calcium, and it presents as worsening hypocalcaemia as the healing skeleton takes up mineral; it is prevented by giving calcium before or with the vitamin D. Hypercalcaemia and hypercalciuria follow over-treatment with vitamin D or calcitriol, particularly in the genetic forms where calcitriol is used long-term, and they are monitored by checking calcium and, where relevant, a urine calcium-to-creatinine ratio. Nephrocalcinosis is a recognised complication of the phosphate therapy used in X-linked hypophosphataemia and is monitored by renal ultrasound. [1] [9]
The diagnostic pitfalls are the ones that misdirect treatment. The first is treating all rickets with vitamin D, which fails in calcium-deficiency rickets, in X-linked hypophosphataemia, and in vitamin D-dependent rickets. The second is dismissing rickets because the 25-hydroxyvitamin D is normal, which misses calcium-deficiency and phosphopenic disease. The third is escalating the vitamin D dose for a persistently high alkaline phosphatase that actually reflects non-adherence or an unrecognised hereditary cause. The fourth is treating hypophosphatasia with bisphosphonates, which worsens it. Each pitfall is avoided by reading the full biochemical pattern before prescribing. [5] [11]
Prognosis & Disposition
The prognosis of nutritional rickets, treated correctly and early, is excellent. The alkaline phosphatase falls within weeks, the biochemistry normalises within months, and the radiograph heals within three to six months, with the deformity often remodelling spontaneously as the child grows. Growth catches up, the motor milestones recover, and the child is left with no residual deficit provided the cause is addressed and supplementation is sustained through adolescence. The hypocalcaemic infant who is resuscitated promptly recovers fully and has no ongoing neurological sequelae attributable to the rickets. [1] [12]
The hereditary forms carry a different prognosis because they require lifelong treatment and surveillance. X-linked hypophosphataemia, treated conventionally or with burosumab, achieves biochemical control and improved growth and deformity, but the child remains short relative to peers and needs multidisciplinary care through childhood, adolescence, and the transition to adult endocrinology. Vitamin D-dependent rickets type 1A responds well to lifelong calcitriol; type 2A is more variable. The prognosis in all of these is framed honestly as a chronic, treatable disease rather than a cure, with the emphasis on adherence, monitoring, and timely orthopaedic intervention. [9] [5]
The disposition follows the cause and the severity. The acutely hypocalcaemic infant is admitted for intravenous calcium and observation. Nutritional rickets that is uncomplicated is managed as an outpatient with a clear plan, a named coordinator, and a defined monitoring schedule, and the public-health arm is activated to screen and treat siblings and the mother. Hereditary rickets is managed jointly with paediatric endocrinology, nephrology, orthopaedics, and dentistry, with genetics involved for counselling. The single most important prognostic step is to sustain the daily 400 IU of vitamin D and an adequate calcium intake through adolescence. [1] [3]
Special Populations
Indigenous children in Australia and Aotearoa New Zealand, and migrant, refugee, and asylum-seeking families, carry a disproportionate burden of nutritional rickets through the combination of darker skin, cultural dress, high-latitude residence, and reduced access to supplementation and follow-up. A fellowship answer names these determinants directly, because they explain the presentation and they direct the response: the treatment is not only cholecalciferol and calcium but also the public-health and primary-care links that make supplementation sustainable. Telehealth and outreach extend specialist surveillance into rural and remote communities where a clinic-based model would fail. [6] [8]
Socioeconomic disadvantage shapes outcome through the practical determinants of adherence rather than through the biology of the disease. A family that cannot reliably obtain the supplement, cannot afford calcium-rich foods, or cannot attend monitoring will see recurrence regardless of the prescription, and the team's job is to remove those barriers through supply, transport, and coordination rather than to escalate the dose. Screening siblings and the mother is standard, because nutritional rickets in one child almost always signals risk in the household. [1] [3]
The preterm infant, the child with cerebral palsy or limited mobility, and the technology-dependent child form further populations in their own right. Preterm infants need supervised mineral and vitamin D supplementation to prevent osteopenia of prematurity; immobile children have reduced skeletal loading and may need attention to weight-bearing and to their mineral balance; and children on long-term anticonvulsants or restricted diets need ongoing surveillance of their vitamin D status. Each population is managed with the same biochemical principles applied to its particular risk profile. [4] [5]
Trauma-informed, interpreter-supported communication is the thread that runs through all of these populations, because the conversation about rickets is often also a conversation about guilt, about cultural practices, and about the safety of sunlight and supplementation. Reassuring a mother that she did not cause her infant's rickets, that exclusive breastfeeding was the right choice, and that the supplement is safe is part of the treatment as much as the cholecalciferol. The recurrence-risk counselling extends to siblings and to future pregnancies. [3] [1]
Evidence, Guidelines & Regional Differences
The foundational evidence is the 2016 Global Consensus Recommendations on the Prevention and Management of Nutritional Rickets, endorsed by the major paediatric endocrine societies, which set the 400 IU daily intake, the 50 nmol per litre sufficiency threshold, and the dual daily-and-stoss treatment regimens. The 2011 Endocrine Society clinical practice guideline on vitamin D deficiency and the 2008 American Academy of Pediatrics statement on the prevention of rickets frame the screening and supplementation practice in North America and elsewhere. Together these documents anchor the operational standards. [1] [2]
The epidemiological and clinical evidence comes from hospital-based series such as the Glasgow cohort, which documented the resurgence of symptomatic vitamin D deficiency and its clinical features in a high-income setting, and from the global reviews that map nutritional rickets across continents and that separate calcium-deficiency from vitamin D-deficiency disease. The 2017 Nature Reviews Disease Primers article on rickets consolidates the pathophysiology and the classification into a single reference, and the 2023 treatment review summarises the contemporary management of vitamin D deficiency in children. [8] [5]
Regional differences are real and shape practice. Australia and New Zealand recommend universal vitamin D supplementation of 400 IU daily for breastfed infants from soon after birth, with particular attention to dark-skinned, veiled, and migrant families, and they extend specialist surveillance through telehealth into rural and remote communities. The United Kingdom, through the Scientific Advisory Committee on Nutrition and the NHS Healthy Start scheme, recommends a daily supplement for all under-fives and targets at-risk groups. North America follows the AAP guidance, and Europe follows the European Society for Paediatric Endocrinology position. Fortification of formula and some foods differs between jurisdictions and explains part of the variation in incidence. [3] [1]
The genuine controversies are few but worth naming. The optimal maintenance dose and the threshold for defining sufficiency are debated at the margins, and the role of routine antenatal vitamin D screening is not uniform. In X-linked hypophosphataemia, the place of burosumab versus conventional phosphate and calcitriol, and the timing of orthopaedic intervention, are evolving. A fellowship answer states the consensus, names the regional variation, and signals where the evidence is still developing rather than asserting false certainty. [9] [12]
[1]Exam Pearls
The five anchors of a fellowship answer are: read the biochemistry before the vitamin D level, separate calcipenic from phosphopenic rickets, give calcium before vitamin D in the hypocalcaemic infant, treat nutritional rickets with cholecalciferol and calcium, and reserve calcitriol with phosphate (or burosumab) for X-linked hypophosphataemia. Hold these five and the topic holds together under any examiner pressure, because they map the recognition, the classification, the emergency, and the two treatment pathways. [1] [5]
The dose anchors are the daily 400 IU for prevention, the 2000 IU daily for three months or the stoss 100 000 to 300 000 IU for treatment of nutritional rickets, and the oral calcium at 30 to 50 mg per kilogram per day elemental given with the vitamin D. The monitoring anchors are a 25-hydroxyvitamin D above 50 nmol per litre for sufficiency, a falling alkaline phosphatase within four to six weeks, and radiographic healing within three to six months. The emergency anchor is intravenous calcium gluconate for the seizing infant, slowly, with cardiac monitoring. [1] [12]
The traps are the mirror image of the anchors. Treating all rickets with vitamin D fails in calcium-deficiency, X-linked hypophosphataemia, and vitamin D-dependent rickets; a normal vitamin D never excludes rickets; a high alkaline phosphatase that does not fall means non-adherence or a hereditary cause, not more vitamin D; and a low alkaline phosphatase in a rachitic child is hypophosphatasia, where bisphosphonates harm. The single most preventable error is giving a stoss vitamin D dose to a hypocalcaemic infant without concurrent calcium and triggering hungry-bone seizures. [5] [10]
References
- [1]Munns CF, Shaw N, Kiely M, et al. Global Consensus Recommendations on Prevention and Management of Nutritional Rickets. J Clin Endocrinol Metab, 2016.PMID 26745253
- [2]Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab, 2011.PMID 21646368
- [3]Wagner CL, Greer FR, American Academy of Pediatrics Section on Breastfeeding and Committee on Nutrition. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics, 2008.PMID 18977996
- [4]Abrams SA, Committee on Nutrition. Calcium and vitamin d requirements of enterally fed preterm infants. Pediatrics, 2013.PMID 23629620
- [5]Carpenter TO, Shaw NJ, Portale AA, et al. Rickets. Nat Rev Dis Primers, 2017.PMID 29265106
- [6]Creo AL, Thacher TD, Pettifor JM, et al. Nutritional rickets around the world: an update. Paediatr Int Child Health, 2017.PMID 27922335
- [7]Prentice A. Nutritional rickets around the world. J Steroid Biochem Mol Biol, 2013.PMID 23220549
- [8]Ahmed SF, Franey C, McDevitt H, et al. Recent trends and clinical features of childhood vitamin D deficiency presenting to a children's hospital in Glasgow. Arch Dis Child, 2011.PMID 20584848
- [9]Kamenický P, Bacchetta J, Baudoin R, et al. X-linked hypophosphataemia. Lancet, 2024.PMID 39181153
- [10]Mornet E. Hypophosphatasia. Orphanet J Rare Dis, 2007.PMID 17916236
- [11]Thacher TD, Fischer PR, Pettifor JM, et al. Calcium-deficiency rickets. Endocr Dev, 2003.PMID 12964429
- [12]Fischer PR, Thacher TD, Pettifor JM, et al. Treatment of vitamin D deficiency in children. Expert Rev Endocrinol Metab, 2023.PMID 37861060