Paeds · endocrinology-diabetes-and-growth
Tall stature and overgrowth syndromes
Also known as tall stature · overgrowth syndrome · gigantism · Sotos syndrome · Beckwith-Wiedemann syndrome · Weaver syndrome · familial tall stature
A fellowship approach to the tall child: separate the familial and constitutional normal variants from the pathological causes — syndromic overgrowth (Sotos, Beckwith-Wiedemann, Weaver, Simpson-Golabi-Behmel, Tatton-Brown-Rahman), growth-hormone excess, and chromosomal or hormonal delay — using a structured dysmorphology and proportion assessment, targeted genetic testing, and syndrome-specific tumour surveillance.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
OVER-GROW — the seven features that demand investigation
Overview & Definition
Tall stature is the mirror image of short stature in statistical terms — a height lying more than two standard deviations above the age- and sex-specific mean — but its clinical meaning is almost the opposite. Where short stature almost always earns an investigation, tall stature usually earns reassurance, because the great majority of tall children are healthy and reflect the genetic endowment of their parents. The skill is therefore not in detecting the tall child but in redistributing the few who carry a pathological cause out of the reassuring majority, and the redistribution rests on a structured assessment rather than on the height number alone. [6]
Familial tall stature is the commonest single cause. A child whose parents are both tall has a high mid-parental target height, grows at a normal velocity along a high centile, has a normal physical examination and body proportions, and a bone age that matches the chronological age. Constitutional advancement of growth is a closely related variant in which an early, self-limited growth spurt and slightly advanced bone age produce a child who is tall relative to peers in mid-childhood but reaches a final adult height within the normal range. Neither of these variants carries intellectual disability, dysmorphism, or tumour risk, and the clinical task is to document the evidence that confirms the variant rather than to investigate reflexively. [7]
The pathological causes demand a different posture, because each carries a consequence that reassurance would delay. Syndromic overgrowth disorders — Sotos, Beckwith-Wiedemann, Weaver, Simpson-Golabi-Behmel, and the Tatton-Brown-Rahman and related methylation-disorder family — pair excess growth with intellectual disability and, in several, a defined embryonal tumour risk. Growth-hormone excess produces gigantism in the open-epiphysis child and carries the cardiovascular and mass consequences of a pituitary adenoma. Chromosomal and hormonal causes, from Klinefelter syndrome to precocious puberty, reshape the growth trajectory and the final height, and several are treatable. The general paediatrician sits at the gate, deciding who is reassured and who is referred. [8] [4]
Classification
The most useful clinical classification sorts the tall child into three buckets, because each bucket drives a different investigation and surveillance plan. The first bucket is the normal variant — familial tall stature and constitutional advancement — which requires no investigation beyond confirmation and which is the destination for most children. The second bucket is endocrine, where an excess or a deficit of a growth-regulating hormone drives the overgrowth: growth-hormone excess, precocious puberty (tall now, short later), and sex-hormone deficiency with delayed epiphyseal fusion. The third bucket is syndromic overgrowth, a genetically heterogeneous family that pairs overgrowth with intellectual disability and often with tumour risk. [6] [8]
The syndromic bucket is where most of the examinable detail lives, and the fellowship candidate should know the characteristic fingerprint of each. Sotos syndrome, caused by haploinsufficiency of the histone methyltransferase NSD1, is the most common syndromic overgrowth disorder, with a characteristic facial gestalt (prominent forehead, receding hairline, pointed chin, long narrow face), advanced bone age, and intellectual disability. Beckwith-Wiedemann syndrome, an imprinting disorder of chromosome 11p15, produces neonatal macrosomia, macroglossia, abdominal-wall defects, and a tumour risk concentrated in the first years of life. Weaver syndrome, caused by EZH2 mutations in the PRC2 complex, pairs overgrowth with a distinctive face, camptodactyly, and intellectual disability. [1] [3]
The remaining syndromic causes are rarer but high-yield. Simpson-Golabi-Behmel syndrome, an X-linked disorder caused by GPC3 loss, produces a coarse face, supernumerary nipples, and a Wilms tumour risk. Tatton-Brown-Rahman syndrome, caused by DNMT3A mutations, produces overgrowth with a round face, mild intellectual disability, and a heightened awareness of haematological malignancy risk. The PTEN hamartoma tumour syndrome family — including Bannayan-Riley-Ruvalcaba syndrome — produces macrocephaly, hamartomas, and a broad tumour spectrum. Marfan syndrome and homocystinuria produce a marfanoid habitus, the former carrying the aortic risk that echocardiography exists to monitor and the latter carrying lens dislocation and thrombosis. [8] [5]

Epidemiology & Risk Factors
Tall stature itself is common, because the normal distribution places roughly two children in every hundred above the ninety-eighth centile. The large majority of these are familial or constitutional variants, which is why the pre-test probability of a pathological cause in a well, proportionate child with tall parents is low. The syndromic overgrowth disorders, by contrast, are individually rare: Sotos syndrome occurs in roughly one in ten to fourteen thousand live births and is the most common of the group, while Beckwith-Wiedemann spectrum occurs in roughly one in ten thousand, and Weaver, Simpson-Golabi-Behmel, and the methylation-disorder family are rarer still. [1] [2]
The risk factors that shift a tall child from the normal-variant bucket into the pathological bucket are clinical rather than epidemiological, and they are the substance of the redistribution assessment. A child with intellectual disability, developmental delay, or a characteristic facial gestalt is at high probability of a syndromic overgrowth disorder. A child with asymmetry, hemihyperplasia, organomegaly, or a neonatal history of macrosomia with an umbilical defect carries the Beckwith-Wiedemann fingerprint and its tumour risk. A child with a marfanoid habitus carries aortic risk, and a child with coarse features and enlarging extremities carries the growth-hormone-excess question. [6] [9]
A family history shapes both the variant and the pathological assessment. Tall parents support familial tall stature, but a family history of sudden cardiac death, aortic disease, or lens dislocation raises the Marfan and homocystinuria questions. A parent with macrocephaly, hamartomas, or multiple primary tumours raises the PTEN hamartoma tumour syndrome question, and a family history of early-onset pituitary tumours raises the AIP and familial isolated pituitary adenoma question in a child with gigantism. The family history is therefore not a box to tick but a set of conditional probabilities that redirect the assessment. [7] [8]
Pathophysiology
Three mechanisms produce excess linear growth, and understanding them is what lets a candidate reason from the bedside to the investigation rather than memorise a list. The first is endocrine excess, in which growth hormone from a pituitary somatotroph adenoma drives hepatic insulin-like growth factor 1 production, and the elevated IGF-1 stimulates both linear growth and soft-tissue proliferation. The pituitary adenomas of childhood gigantism may be sporadic or part of a genetic syndrome — AIP mutations, McCune-Albright, Carney complex, multiple endocrine neoplasia type 1, and the X-linked acrogigantism syndrome driven by GPR3 duplications on Xq26.3, which produces aggressive early-onset gigantism in male infants. [7]
The second mechanism is chromosomal or hormonal, in which the timing of sex-steroid exposure determines the trajectory rather than the growth-hormone axis itself. In Klinefelter syndrome and in delayed or absent puberty, the lack of sex steroids delays epiphyseal fusion, and the child continues to grow into a tall, eunuchoid habitus with arm span exceeding height. In precocious puberty the mechanism is the mirror image: early oestrogen and testosterone drive a brisk early growth spurt that places the child tall relative to peers, but the same hormones fuse the epiphyses early, and the final adult height is below the genetic potential. This tall-now-short-later paradox is a classic fellowship trap, and the recognition that early tallness can be a warning rather than a reassurance is the teaching point. [6] [7]
The third mechanism is the loss of growth-braking genes, which is the unifying theme of the syndromic overgrowth disorders. Many of these genes are epigenetic regulators — histone methyltransferases and DNA methyltransferases — whose normal function is to restrain proliferation by shaping chromatin. NSD1 loss in Sotos syndrome, EZH2 loss in Weaver syndrome through the PRC2 complex, and the 11p15 imprinting defects of Beckwith-Wiedemann that dysregulate IGF-2 and the cell-cycle brake CDKN1C each remove a restraint on growth. The shared consequence is overgrowth paired with intellectual disability, because the same genes that restrain body growth also shape cortical development, and in several the consequence includes tumour risk, because loss of a cell-cycle brake permits embryonal neoplasia. [4] [8]

Clinical Presentation
The presentation of the tall child is shaped by which bucket the child sits in, and a fellowship answer earns depth by separating the normal-variant presentation from the syndromic, the endocrine, and the chromosomal. The normal-variant child is brought by parents concerned about height or by a school that has flagged the child as unusually tall, and the history and examination confirm a well, proportionate child on a stable high centile with tall parents. There is no dysmorphism, no intellectual disability, and no pubertal abnormality, and the reassurance is the intervention. [6]
The syndromic overgrowth disorders declare themselves through a recognisable fingerprint that the history and examination are designed to detect. Sotos syndrome presents in the neonate and infant with excess birth length and head circumference, early rapid growth, advanced bone age, a characteristic facial gestalt of a prominent forehead, receding hairline, pointed chin, and long narrow face, and developmental delay that evolves into mild-to-moderate intellectual disability. The child is often clumsy and hypotonic, and behavioural features including autism-spectrum characteristics and anxiety emerge in childhood. [1]
Beckwith-Wiedemann spectrum presents most recognisably in the neonate with macrosomia, macroglossia that interferes with feeding and breathing, and an anterior abdominal-wall defect ranging from a severe omphalocele to a mild umbilical hernia or diastasis recti. Hemihyperplasia — asymmetric overgrowth of one or more body regions — is a cardinal feature because it marks the child who carries the highest embryonal tumour risk, and the tumour may be the presenting feature when an abdominal mass is found at a routine review. Neonatal hypoglycaemia from islet-cell hyperplasia is common and demands early management. [2] [9]
Growth-hormone excess in the child with open epiphyses produces gigantism rather than the acromegalic picture seen in adults, because the growth plates have not yet fused. The presentation is of a child growing at an excessive velocity with coarsening facial features, enlarging hands and feet, prognathism, and headaches or visual disturbance from the pituitary mass. The marfanoid habitus of Marfan syndrome and homocystinuria presents as a tall, thin child with long limbs and digits, and the discriminating features — lens dislocation and thrombosis in homocystinuria, aortic root dilatation and the characteristic skeletal and skin features in Marfan — are what the assessment is built to find. [5] [7]
Differential Diagnosis
The differential splits into two practical questions: which syndrome is this, and could the tall stature be iatrogenic or secondary. Among the syndromic causes, the discriminating features separate the overgrowth disorders efficiently. Sotos syndrome pairs its characteristic face and advanced bone age with intellectual disability, while Weaver syndrome adds camptodactyly and a different facial gestalt. Beckwith-Wiedemann is set apart by its macrosomia, macroglossia, and abdominal-wall defect, and Simpson-Golabi-Behmel by its coarse face, supernumerary nipples, and X-linked inheritance. The methylation-disorder family — Tatton-Brown-Rahman, Malan, and the related NSD1-spectrum and histone-modifier disorders — is distinguished by overgrowth with a round face and variable intellectual disability, and increasingly by an epigenetic-signature test rather than by bedside features alone. [8] [4]
The marfanoid habitus is its own differential, because several conditions produce a tall, thin child with long limbs. Marfan syndrome is the archetype, with its fibrillin-1 root, aortic root dilatation, lens dislocation that is classically upward, and the revised Ghent criteria that weigh the systemic score. Homocystinuria is the classic metabolic mimic, with downward lens dislocation, intellectual disability, and a thrombosis risk, and it is excluded by plasma homocysteine and amino acids. The Loeys-Dietz syndrome and the Shprintzen-Goldberg syndrome are rarer relatives, and congenital contractural arachnodactyly enters the differential when joint contractures accompany the marfanoid build. [5]
The endocrine and chromosomal differentials are more targeted. Pituitary gigantism is separated from familial tall stature by the elevated IGF-1, the failure to suppress growth hormone on glucose loading, and the pituitary lesion on magnetic resonance imaging. Klinefelter syndrome presents with a tall eunuchoid habitus, small firm testes, and learning difficulty, and is confirmed by karyotype. Precocious puberty produces early tallness with accelerated bone age and signs of sex-steroid excess, and the recognition that the child will be short at final height is what triggers the gonadotropin-releasing-hormone-analogue decision. Obesity can produce apparent tall stature in early childhood with an advanced bone age that resolves toward average in later years, and exogenous oestrogen or androgen exposure must be asked about directly. [6] [7]
| Feature | Sotos (NSD1) | Beckwith-Wiedemann (11p15) | Weaver (EZH2) | Simpson-Golabi-Behmel (GPC3) |
|---|---|---|---|---|
| Birth size | Large, macrocephaly | Macrosomic | Large | Large, macrosomia |
| Face | Prominent forehead, long face | Macroglossia, midface hypoplasia | Broad forehead, hypertelorism | Coarse, macrocephaly |
| Hands | Large, clumsy | Normal | Camptodactyly | Postaxial polydactyly |
| Tumour risk | Low | Wilms, hepatoblastoma | Low | Wilms tumour |
| Inheritance | AD, mostly de novo | Imprinting, complex | AD, de novo | X-linked recessive |
Clinical & Bedside Assessment
The assessment is the hinge of the topic, because the decision to reassure or to investigate rests entirely on what the bedside examination finds. Begin with the growth data: plot the height, the weight, and the head circumference accurately on the appropriate reference chart, calculate the mid-parental target height and its range, and measure the growth velocity over at least two points separated by six months. A child who is tall and growing at a normal velocity on a stable centile with tall parents is a normal variant until proven otherwise, but a child whose height is crossing centiles upward has a velocity problem and must be investigated regardless of how well the child looks. [6] [7]
The physical examination is a structured hunt for the redistributors. Measure the body proportions — the upper-to-lower segment ratio and the arm span against height — because eunuchoid proportions (arm span exceeding height by more than five centimetres) redirect toward Klinefelter and delayed puberty, and a reduced upper-to-lower ratio accompanies the marfanoid habitus. Examine the face for the characteristic gestalt of each overgrowth syndrome, the hands for camptodactyly or postaxial polydactyly, the abdomen for organomegaly or an abdominal-wall defect, and the skin for cafe-au-lait patches that raise the McCune-Albright and NF1 questions. Look actively for asymmetry and hemihyperplasia, because the asymmetry is the sign that triggers the tumour surveillance. [8] [2]
Pubertal staging is mandatory, because both the timing and the tempo redirect the assessment. Precocious puberty — the appearance of secondary sexual characteristics before the age-bound thresholds — produces early tallness with advanced bone age, and the recognition that the final height will be compromised is what drives the treatment decision. Delayed or absent puberty with eunuchoid proportions raises Klinefelter and constitutional delay, and the karyotype and the gonadotropin profile separate them. In the infant, examine the tongue, the umbilicus, and the size and symmetry of the body for the Beckwith-Wiedemann fingerprint, and check for neonatal hypoglycaemia that may need ongoing management. [6] [9]
Development and cognition close the assessment, because the pairing of overgrowth with intellectual disability is the hallmark of the syndromic bucket. A tall child with normal development, normal proportions, and a normal examination is a normal variant, but a tall child with developmental delay, learning difficulty, or behavioural features earns a genetic investigation regardless of the height trajectory. The family's practical context matters as much as the clinical detail: the height of each parent and sibling, the family history of tumours, sudden death, and aortic disease, and the psychosocial burden that tall stature may impose on a child at school. Combining the growth data with the physical and developmental examination is what produces the redistribute-or-reassure decision. [10] [1]
Investigations
The investigation plan follows the assessment, and the first principle is that a well, proportionate child with tall parents and a normal velocity needs no investigation beyond a bone age for confirmation. A bone age radiograph of the left hand and wrist is the single test that earns its place in the normal-variant child, because an age-matched bone age confirms familial tall stature while an advanced bone age redirects toward constitutional advancement or precocious puberty. Over-investigating a reassuring child is a common error, and a clear documentation of the variant evidence is the safeguard. [6]
When the assessment redistributes toward endocrine pathology, the investigations are targeted to the suspected axis. A child with coarse features, enlarging extremities, or a height crossing centiles upward earns a serum IGF-1 and IGFBP-3, and if these are elevated, an oral glucose suppression test and a pituitary magnetic resonance imaging scan to confirm and localise the growth-hormone-secreting adenoma. A child with signs of precocious puberty earns a bone age, a gonadotropin and sex-steroid profile, and where indicated a stimulation test and imaging. A child with eunuchoid proportions earns a karyotype for Klinefelter syndrome and a gonadotropin profile to characterise the pubertal delay. The endocrine investigation is therefore axis-specific and not a broad screen. [7]
When the assessment redistributes toward syndromic overgrowth, the genetic investigation has become phenotype- and epigenetic-signature driven. A child with the Sotos facial gestalt and developmental delay earns NSD1 testing — deletion or sequencing — first-line. A child with the Beckwith-Wiedemann fingerprint earns 11p15 imprinting testing, which includes methylation analysis of the two imprinting centres and copy-number analysis, because the molecular subgroup determines the tumour risk and the surveillance intensity. A child with overgrowth and intellectual disability of no obvious cause earns a chromosomal microarray and an epigenetic overgrowth panel, and increasingly a methylation-signature test that can classify several overgrowth syndromes from a single assay. [2] [8]
Tumour surveillance is an investigation in its own right, and it is the consequence of the Beckwith-Wiedemann and Simpson-Golabi-Behmel diagnoses. A child with hemihyperplasia or a molecular subtype carrying tumour risk earns an abdominal ultrasound every three months through the first years of life and at least until age eight, directed at Wilms tumour and hepatoblastoma, alongside periodic serum alpha-fetoprotein for hepatoblastoma detection. Marfan syndrome earns a baseline and annual echocardiogram for the aortic root. The genetic and the surveillance investigations are therefore linked: the molecular diagnosis dictates the surveillance schedule, and the surveillance is what prevents the preventable death. [9] [12]
Management — Resuscitation
Tall stature is rarely a resuscitation-grade problem, but several of its causes declare themselves with acute neonatal or childhood threats that demand immediate recognition. The neonate with Beckwith-Wiedemann spectrum may present with macroglossia severe enough to obstruct the airway, with a large omphalocele that demands surgical and fluid management, and with neonatal hypoglycaemia from pancreatic islet-cell hyperplasia that requires prompt glucose measurement and intravenous dextrose. The hypoglycaemia can be severe and refractory, and it is the immediate neonatal threat that the paediatric team must anticipate. [2] [12]
Growth-hormone excess can present with an acute pituitary apoplexy or with visual loss and headache from mass effect, and the recognition that a rapidly growing child with coarsening features may harbour a pituitary macroadenoma is what drives the urgent imaging and the endocrine referral. Marfan syndrome presents its acute threat as aortic dissection, which is rare in young children but rises through adolescence and young adulthood, and the baseline and serial echocardiography that the diagnosis triggers are the resuscitation-grade prevention. Homocystinuria presents its acute threat as venous or arterial thrombosis, including perioperatively, and the avoidance of unnecessary surgery and the perioperative thromboprophylaxis are part of the acute management. [5]
The psychosocial and safeguarding dimension is a resuscitation of a different kind. A child whose height is causing significant distress — bullying, social isolation, or the practical burden of being far larger than peers — may present with anxiety or school refusal, and the recognition that the psychosocial burden is itself a treatable problem is part of the management. Equally, the finding of an unrecognised overgrowth syndrome in a child whose intellectual disability or tumour risk has been missed is a safeguarding-grade error, and the early establishment of the diagnosis and the surveillance is the corrective. [10]
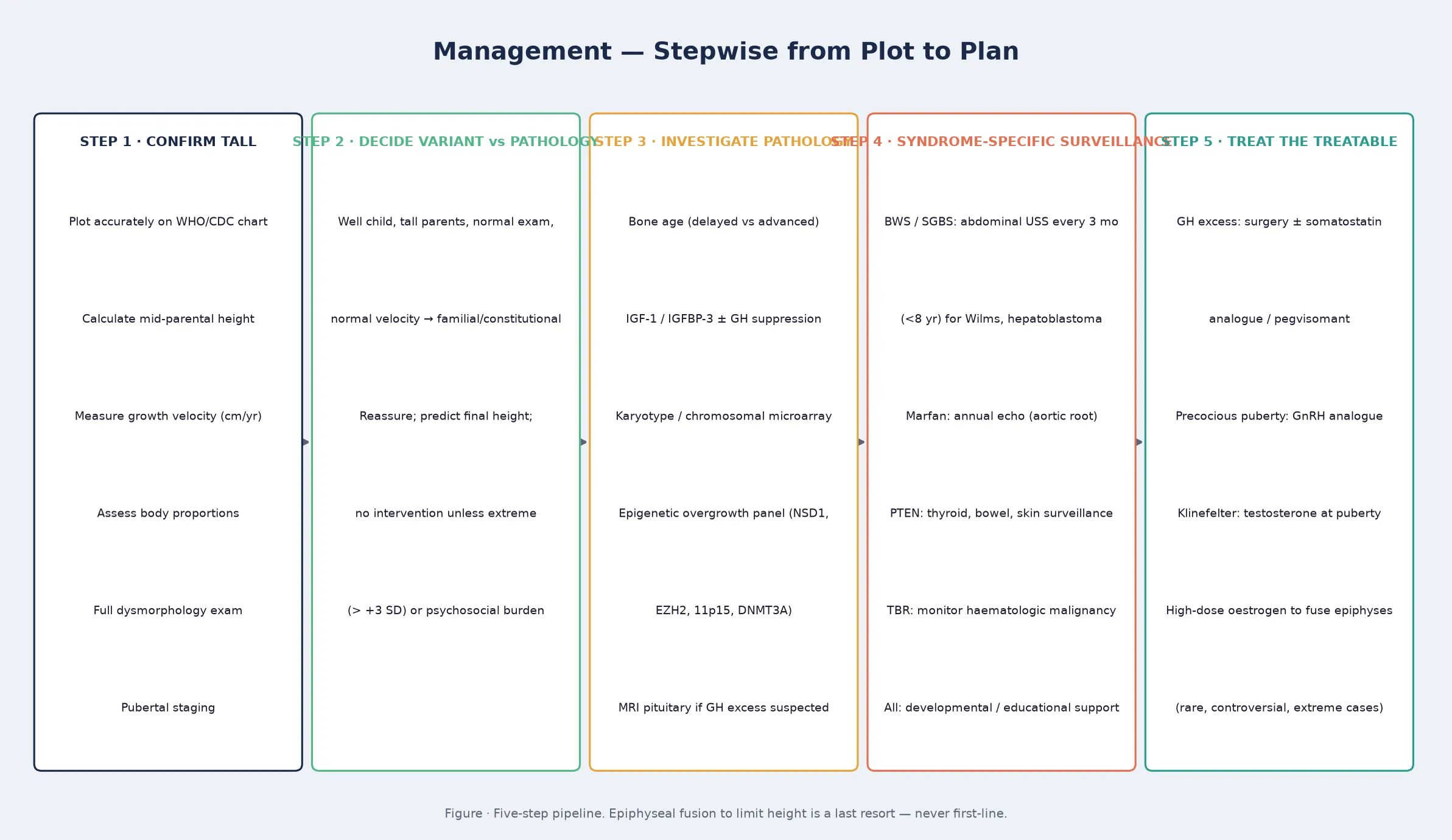
Management — Definitive & Stepwise
Definitive management of the tall child begins with the reassurance of the normal variant, because most tall children need no treatment. The prediction of the final adult height, using the bone age and the mid-parental target, is the intervention, and the documentation of the reassurance in the record is what protects the family from unnecessary referral and the child from unnecessary testing. The temptation to intervene pharmacologically to limit height in a normal-variant child should be resisted, because the high-dose sex-steroid therapy once used to fuse the epiphyses carries thrombotic, oncologic, and psychosocial risks that outweigh the benefit in all but the most extreme cases, and contemporary practice reserves it for rare situations of projected extreme final height with a heavy psychosocial burden. [6] [7]
For the endocrine causes, the management is cause-specific and often curative. Pituitary gigantism is managed by a specialist pituitary team using transsphenoidal surgery as first-line, with somatostatin analogues, growth-hormone receptor antagonists (pegvisomant), and dopamine agonists as medical therapy when surgery is incomplete or the tumour recurs. Precocious puberty is managed with a gonadotropin-releasing-hormone analogue to suppress the hypothalamic-pituitary-gonadal axis, preserve final height, and address the psychosocial burden, and the treatment is monitored with growth velocity, bone age, and sex-steroid levels. Klinefelter syndrome is managed with testosterone replacement from adolescence to induce secondary sexual characteristics and preserve bone and metabolic health. [7]
For the syndromic overgrowth disorders, the management is a syndrome-specific surveillance and support plan rather than a growth-modifying therapy, because the overgrowth itself is not treatable. Sotos syndrome management centres on developmental and educational support, behavioural intervention for the autism-spectrum and anxiety features, and routine medical surveillance. Weaver and the methylation-disorder family are managed similarly, with the developmental support tailored to the intellectual profile. Beckwith-Wiedemann management is dominated by the neonatal problems — airway, abdominal wall, and hypoglycaemia — and by the tumour surveillance that follows the molecular diagnosis, and the closure of the abdominal-wall defect and the reduction of the macroglossia are surgical decisions made in the first years. [1] [2]
Specific Subtypes & Scenarios
Beckwith-Wiedemann spectrum is the subtype that carries the most operational detail, because its molecular subgroups drive different tumour risks and surveillance intensities. The 11p15 region harbours two imprinting centres, and the loss of methylation on the maternal allele at imprinting centre 2 is the commonest molecular finding and carries a moderate tumour risk, while gain of methylation at imprinting centre 1 and paternal uniparental disomy carry higher risks, and the CDKN1C loss-of-function mutation and the cytogenetic abnormalities carry their own profiles. The fellowship candidate who can connect the molecular subgroup to the tumour risk and the surveillance plan demonstrates a mature command of the condition. [2] [9]
Sotos syndrome is the commonest syndromic overgrowth disorder and carries its own constellation of management issues. The characteristic facial gestalt becomes more recognisable with age, the advanced bone age contributes to early tallness, and the intellectual disability is typically mild to moderate with a variable behavioural phenotype that includes autism-spectrum characteristics, anxiety, and behavioural rigidity. The tumour risk is lower than in Beckwith-Wiedemann but is not absent, and several surveillance schedules include periodic screening for the small number of associated neoplasms. The management is developmental and educational support, behavioural intervention, and the routine surveillance that the GeneReviews schedule sets out. [1]
Growth-hormone excess in childhood — gigantism — is a distinct scenario that demands a pituitary specialist. The X-linked acrogigantism syndrome, caused by duplications of the GPR3 gene in the Xq26.3 region, produces a uniquely aggressive early-onset gigantism that begins in infancy and is overwhelmingly seen in males, and the recognition of this specific entity matters because it demands intensive multimodal therapy and carries a guarded prognosis. The familial isolated pituitary adenoma syndrome, driven by AIP mutations, presents with gigantism or acromegaly in children and young adults and warrants family screening. [7]
The marfanoid child is a scenario that crosses endocrinology, genetics, and cardiology. Marfan syndrome, confirmed against the revised Ghent nosology using the systemic score, the echocardiographic aortic dimensions, and the lens examination, is managed with serial echocardiography, beta-blocker or angiotensin-receptor-blocker therapy to protect the aortic root, and surgical aortic repair when the root reaches a threshold dimension. Homocystinuria, the metabolic mimic, is managed with pyridoxine, folate, and betaine to lower the homocysteine, a methionine-restricted diet, and thromboprophylaxis. The two conditions are distinguished by the direction of the lens dislocation and by the plasma homocysteine, and the distinction matters because the management and the prognosis diverge. [5]
Why molecular subgroup drives the Beckwith-Wiedemann tumour plan
The tumour risk in Beckwith-Wiedemann spectrum is not uniform — it depends on the molecular subgroup, because different imprinting defects dysregulate different cell-cycle brakes. The loss of methylation at imprinting centre 2, the commonest finding, carries a moderate tumour risk that warrants surveillance. Paternal uniparental disomy and the gain of methylation at imprinting centre 1 carry higher risks that demand the most intensive abdominal ultrasound schedule. CDKN1C mutations and the cytogenetic abnormalities carry their own profiles. The consequence is that the molecular result dictates the surveillance intensity: a child cannot be safely placed on a tumour surveillance schedule until the 11p15 molecular subgroup is known, and ordering the imprinting analysis at diagnosis is the step that makes the surveillance evidence-based. [2] [9]
Complications & Pitfalls
The harm in tall stature comes most often from the missed pathological diagnosis — the child reassured as "just tall" who carries a Beckwith-Wiedemann tumour risk, a Marfan aortic risk, or an unrecognised growth-hormone excess — and the mechanism of the miss is consistent. The height is plotted, the child looks well, and the dysmorphology, the proportions, the asymmetry, and the developmental assessment are never completed. The cardinal pitfall is therefore the reassurance that precedes the assessment, and the safeguard is the structured redistribution that the OVER-GROW mnemonic is designed to drive. [6] [8]
The emergent complications that must not be missed cluster in the neonate and the older child. Neonatal hypoglycaemia from Beckwith-Wiedemann islet-cell hyperplasia can cause seizures and long-term neurodevelopmental harm if not anticipated and managed. Airway obstruction from severe macroglossia, and the surgical and fluid challenges of a large omphalocele, are the neonatal surgical threats. The abdominal embryonal tumours — Wilms tumour and hepatoblastoma — declare themselves most often in the first years, and the child on a surveillance schedule is the child whose tumour is found when it is still treatable. Aortic dissection in Marfan syndrome is the catastrophe that serial echocardiography exists to prevent, and thrombosis in homocystinuria is the catastrophe that biochemical management exists to prevent. [2] [5]
The management pitfalls share a common root: treating or reassuring before the assessment is complete. Overtreating a normal-variant child with high-dose sex steroids to limit height is a pitfall that contemporary practice has largely retired but that still appears when the psychosocial pressure on the family is intense. Missing the molecular subgroup in Beckwith-Wiedemann, and therefore placing the child on the wrong surveillance schedule or none at all, is the preventable error that costs lives. Confusing Marfan and homocystinuria, and treating the aorta when the homocysteine is the threat, is the diagnostic pitfall that the lens examination and the biochemistry are designed to resolve. [9] [5]
Prognosis & Disposition
The prognosis of the tall child is determined entirely by the cause. The normal-variant child has an excellent prognosis: the final height is predicted from the bone age and the mid-parental target, the health is normal, and the psychosocial outcome depends on the support the child and family receive rather than on any medical intervention. The endocrine causes carry a prognosis determined by the treatability of the underlying condition — pituitary gigantism is curable with surgery and medical therapy in many cases, precocious puberty is manageable with a GnRH analogue, and Klinefelter syndrome is managed with testosterone and fertility considerations through adolescence and adulthood. [7]
The syndromic overgrowth disorders carry a prognosis shaped by the intellectual disability, the tumour risk, and the syndrome-specific medical problems, and the single biggest determinant of outcome is the timing and the consistency of the surveillance. A child with Beckwith-Wiedemann spectrum whose tumour surveillance is consistent has a tumour-related outcome that is dramatically better than the child whose surveillance lapses, and a child with Marfan syndrome whose aortic surveillance is consistent has a cardiovascular outcome that is dramatically better than the child whose echocardiography is missed. The prognosis is therefore a function of the match between the surveillance and the risk, and the plan is built around that match. [9] [12]
The general paediatrician owns the gate and the coordination, and the disposition is shared, structured care. The endocrinologist owns the growth-hormone-excess and the pubertal-disorder management, the clinical geneticist owns the molecular diagnosis and the counselling, the oncologist and the surgeon own the tumour management, the cardiologist owns the aortic surveillance, and the developmental and educational teams own the intellectual-disability support. A named coordinator prevents the fragmentation that is the enemy of a checklist-based plan, and the referral to a specialist multidisciplinary service at the point of diagnosis — rather than after a complication — is what measurably improves the outcome. [10] [8]
Special Populations
The overgrowth syndromes interact with the child's social, cultural, and developmental context, and the same surveillance plan behaves differently across populations — access, adherence, and late presentation each shape the outcome. A plan that is clinically correct but unattainable for a family is no plan at all, and the fellowship answer recognises that the schedule is only as good as the family's ability to engage with it. The tumour surveillance schedule, in particular, depends on reliable attendance for abdominal ultrasound every three months, and any barrier to that attendance is a barrier to the outcome the surveillance exists to protect. [9] [12]
Indigenous children in Australia and New Zealand may carry a higher background burden of the conditions that complicate overgrowth — renal disease, metabolic disease, and the social determinants that affect access to specialist care — and the reduced access to specialist and imaging services in remote communities intensifies the need for early, structured surveillance and for a low threshold to investigate symptoms. Telehealth and outreach extend the surveillance net into communities that a clinic-based model would miss, and the local primary-care team is the asset that holds the plan together when the specialist service is distant. [6]
Migrant, refugee, and asylum-seeking families may arrive with incomplete growth records, an uncertain family history, and no prior genetic testing, and the diagnosis of an overgrowth syndrome may not have been made in the country of origin. A careful reconstruction of the growth trajectory from any available records, confirmation of the molecular diagnosis and its inheritance, and an interpreter-mediated explanation of the surveillance plan are the foundations. The written schedule is provided in the family's language, and the tumour surveillance is reconciled with the family's mobility and the local service availability. [10]
The school-age child with intellectual disability and an overgrowth syndrome is a population in their own right, because the uneven developmental profile is easily misread as behavioural difficulty or as global delay. Targeted educational and psychosocial support, tailored to the intellectual profile and the behavioural phenotype, is the intervention, and trauma-informed, strengths-based communication extends to the genetic-counselling conversation itself. Socioeconomic disadvantage shapes late presentation and care-coordination feasibility, because the limiting step is often attendance and transport rather than the medicine, and structuring the surveillance around a single coordinated visit improves engagement. [1] [8]
Evidence, Guidelines & Regional Differences
The evidence base for the syndromic overgrowth disorders has been reshaped by the discovery that the genes involved are overwhelmingly epigenetic regulators, and the 2017 Tatton-Brown Childhood Overgrowth Consortium study established that mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability, reframing the field from a collection of named syndromes to a biology of chromatin dysregulation. The 2024 Lui and Baron review in the Journal of Clinical Endocrinology and Metabolism synthesises the epigenetic causes of overgrowth syndromes and is the current mechanistic reference. The Argente 2019 review in Frontiers in Endocrinology frames the practical question of which growth-disordered patients require genetic testing. [8] [4] [10]
The operational guidance for the individual syndromes rests on consensus documents and registries. The 2018 Brioude expert consensus in Nature Reviews Endocrinology is the foundational document for Beckwith-Wiedemann spectrum, setting out the clinical and molecular diagnosis, the screening, and the management, and it is updated by the 2025 Russo Italian register update and by the Maas 2016 subgroup-specific tumour-risk data. Sotos syndrome is comprehensively covered in the GeneReviews chapter by Adam and colleagues, which is periodically updated, and Weaver syndrome is anchored by the 2012 Gibson EZH2 discovery paper. Marfan syndrome is governed by the 2010 revised Ghent nosology of Loeys, which remains the diagnostic standard. [2] [1] [3] [5] [12]
Where the evidence is weak, a fellowship answer says so honestly. The role of height-limiting therapy with high-dose sex steroids has diminished as the risks have become clearer, and contemporary practice reserves it for rare extreme cases, but the evidence base for its risk-benefit balance is thin and contested. The optimal frequency and duration of tumour surveillance in the lower-risk Beckwith-Wiedemann subgroups, and the tumour risk in several of the rarer methylation-disorder syndromes, are areas of genuine uncertainty, and regional practices differ in the availability of methylation-signature testing and in the specialist multidisciplinary services. Naming the uncertainty is a mark of intellectual honesty that examiners reward. [6] [9]
In Australia and New Zealand, the evaluation of the tall child follows the international consensus, with growth plotted on the WHO or CDC reference charts and the redistribution assessment driving the investigation. Genetic testing — chromosomal microarray, NSD1 testing for Sotos, and the 11p15 imprinting analysis for Beckwith-Wiedemann — is accessed through the state clinical genetics services and the specialist paediatric endocrinology centres, and the epigenetic-signature testing that classifies several overgrowth syndromes from a single assay is increasingly available. Tumour surveillance for the Beckwith-Wiedemann and hemihyperplasia children operates through the paediatric oncology services, with abdominal ultrasound accessed through regional imaging. Access to specialist genetics, endocrinology, and oncology services is uneven across rural and remote communities, which intensifies the need for early referral, telehealth-supported surveillance, and a coordinator who holds the plan. [2] [6]
Exam Pearls
A fellowship candidate answering on tall stature and overgrowth syndromes should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for; the traps are where easy marks are lost. [6] [10]
Anchor one: redistribute, do not just reassure. Every tall child earns a structured assessment — proportions, dysmorphology, asymmetry, puberty, and development — before being labelled familial. The OVER-GROW mnemonic drives the redistribution, and any positive feature redirects the child from reassurance to investigation. [6]
Anchor two: know the three mechanism-buckets. Endocrine excess (growth-hormone-driven gigantism), chromosomal and hormonal timing (Klinefelter, delayed and precocious puberty), and syndromic overgrowth (the epigenetic and single-gene family) are the three routes to tall stature, and reasoning from the bedside to the bucket is the skill. [7]
Anchor three: the syndromic overgrowth fingerprint. Sotos (NSD1, characteristic face, intellectual disability), Beckwith-Wiedemann (11p15, macrosomia, macroglossia, abdominal wall, tumour risk), Weaver (EZH2, camptodactyly), Simpson-Golabi-Behmel (GPC3, coarse, X-linked), and Tatton-Brown-Rahman (DNMT3A, overgrowth with intellectual disability) are the named syndromes the candidate holds. [1] [8]
Anchor four: tumour surveillance is dictated by the molecular subgroup. In Beckwith-Wiedemann spectrum and in hemihyperplasia, the 11p15 molecular subgroup determines the tumour risk and the abdominal-ultrasound schedule — every three months until age eight for the higher-risk subgroups. The molecular result is the prerequisite for an evidence-based surveillance plan. [2] [9]
Anchor five: treat the treatable, reserve height-limiting therapy for the extreme. Pituitary gigantism is curable, precocious puberty is manageable, and Klinefelter is treatable — these are the interventions that change the trajectory. High-dose sex steroids to limit height in a normal-variant child is a last resort, not a first-line option, and contemporary practice reserves it for rare extreme cases. [7] [6]
The three traps to avoid are reassuring before assessing, placing a Beckwith-Wiedemann child on tumour surveillance without knowing the molecular subgroup, and confusing Marfan with homocystinuria. Sotos is the commonest syndromic overgrowth disorder, Beckwith-Wiedemann carries the tumour risk, NSD1 and EZH2 and DNMT3A and the 11p15 locus are the high-yield genes, and the revised Ghent nosology governs Marfan — the high-yield facts a candidate holds. Avoid the traps and land the anchors, and the answer falls into place. [8] [5]
References
- [1]Adam MP, Bick S, Mirzaa GM, Otero FJ, Toriello HV, Tatton-Brown K. Sotos Syndrome. GeneReviews, 1993.PMID 20301652
- [2]Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, Boonen SE, Cole T, Baker R, Bertoletti M, Cocchi G, Coze C, De Pellegrin M, Filipovic-Grcic M, Gebauer J, Heide S, Hyland EMJ, Linglart A, Maas SM, Macdonald F, Pèrez de Nanclares G, Rusu C, Sá J, Setó A, Skórka A, Tønnesen H, Tümer Z, Aris J, de Pagter MS, Lapunzina P, Prawitt D, Netchine I, Hennekam RC, Weksberg R, Alders M, Eggermann T, Mackay DJG, Riccio A, Maher ER, Mannens MM. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome. Nat Rev Endocrinol, 2018.PMID 29377879
- [3]Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, Marra MA; FORGE Canada Consortium, Chai DH, McLeod DR, Fernandes NF, Lorenz S, Reis A, Hewson S, Choufani S, Weksberg R, Tétreault M, Awadalla P, Osio K, Tsui LC, Robinson WP, Rosenberg C, Marshal CR. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet, 2012.PMID 22177091
- [4]Lui JC, Baron J. Epigenetic Causes of Overgrowth Syndromes. J Clin Endocrinol Metab, 2024.PMID 37450557
- [5]Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet, 2010.PMID 20591885
- [6]Caro R, Savel P, Moss PI. Evaluation of Short and Tall Stature in Children. Am Fam Physician, 2025.PMID 40531152
- [7]Sada V, Puliani G, Feola T, Gianfrilli D, Lenzi A, Radicioni AF, Isidori AM. Tall stature and gigantism in transition age: clinical and genetic aspects-a literature review. J Endocrinol Invest, 2024.PMID 37891382
- [8]Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, Elliott A, Wylie H, Ardissone A, Rittinger O, Stewart F, Temple IK, Cole T; Childhood Overgrowth Consortium, Mahamdallie S, Seal S, Ruark E, Rahman N. Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability. Am J Hum Genet, 2017.PMID 28475857
- [9]Maas SM, Vansenne F, Kadouch DJ, Ibrahim A, Bliek J, Hopman S, Mannens MM, Merks JH, Maher ER, Hennekam RC. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A, 2016.PMID 27419809
- [10]Argente J, Tatton-Brown K, Lehwalder D, Pfäffle R. Genetics of Growth Disorders-Which Patients Require Genetic Testing? Front Endocrinol (Lausanne), 2019.PMID 31555216
- [11]Shen W, Heeley JM, Carlston CM, Rose NC, Haverfield EV, Tamburrino MB, Nelson CC, Newkirk HL, Smith JL, Erikson K, Sheng X, Smith R, Sankar R, Tsai AC, Schulz J, Shinawi M, Surti U, Bellus G, Ennis Y, O'Sullivan MJ, Willaert R, Rajan-Babu IS, Rao S, Kassai M, Mohnach L, Lopes FD, Seawright M, Mancini EMS, Mantovani G, de Nanclares GP, Lapunzina P, Hisado-Oliva A, Garin I, Argente J, López-Siguero JP, Campos-Barros A, Morgan T, Li D, Zhang VW, Wong LJ, Bhoj EJ, Zackai EH, Spinner NB, Krantz ID, Romero CJ, Parodisi R, Wenzel W, Bick D, Hanchard N, Carmany EP, Stemmerhoff L, Watts A, Murthy C, Vasquez-Velasquez C, Durand C, Mulder PA, Hennekam RC, Foster A, Magoulas PL, Du H, Davenport ML, Hogan SR, Gracia R, Madan-Khetarpal R, Champaigne J, McGuire M, Bhatt R, Zambrano RM, Carmona-Aldana F, Camacho CA, Kaur M, Martin JA, de Tar M, Richardson CE, Raskin S, Schwartz C, Broczek-Buzelin K, Sigaudy S, Missirian C, Philip N, Lerman-Sagie T, Hoss-Feig S, Lacassie Y, Groos Heidenreich S, Fisher J, Hogue J, Hoxha R, Bostwick B, Scott DA, Scotchman E, Patel A, Temple IK, Courtens W, Kini A, Sharma N, Caluseriu O, Bernstein JA, Glass IA, Avela K, Hietala M, Suomi V, Maenpaa H, Shannon N, Abu-Amero S, Blair EM, Pollard RT, Riehme C, Touraine P, Choucair N, Duffourd Y, Thauvin-Robinet C, Faivre L, Lui JC, Baron J, Pérez-Jurado LA, Cueto-González AM, Quesada-López C, Arnedo M, Heine-Súñer D, Wright M, Baynam G, Kiraly-Borri C, Hawkins R, Nicholl J, Campbell I, Dudding T, Bratkovic D, Vijay S, Genêt S, Malan V, Goldenberg A, Giuliano F, Dieux-Coeslier A, Vincent-Delorme C, Désir J, Maystadt I, Debeer A, Devriendt K, Brock P, Wakeling E, Stewart H, Chandler K, Kerr B, Donaldson A, Cole T, Tatton-Brown K, Harbison MD, Hu JC, Guo MH, Inoue T, Higashi N, Hyodo T, Sato T, Yamamoto T, Inagaki T, Numabe H, Tanaka T, Ogata T, Fukami M, Dateki S, Mizuno S, Muroya K, Hasegawa Y, Ihara K, Hara-Ishii C, Tsuboi J, Hattori A, Saida K, Kikuchi-Ueda T, Abe M, Tanaka M, Maeda M, Uehara T, Miyake N, Matsumoto N, Mizuguchi T, Takahashi H, Sawai H, Kosho T, Yamamoto-Shimojima K, Yamamoto T, Tanaka T, Saito Y, Yatsuga S, Sato H, Nakamura A, Sato R, Katoh Y, Miyata I, Yokoi K, Iwama S, Sugihara M, Seo-Mayer T, Beek G, Shealy A, Bassetti JA, Day-Salvatore D, Mendoza-Londono R, Keng J, Brunetti-Pierri N, Peluso P, Morrill A, Tosi L, Wierenga K, Jobanputra V, Murthy C, Wattendorf DJ, Eberhardt Y, Ellard S, Flanagan SE, Mackay DJG, Harbison MD, Bhoj EJ, Deardorff MA, Dormans JP, Bernier FP, Innes AM, Parboosingh JS, Loredo-Osti JC, Casselman M, Dyment DA, FORGE Canada Consortium, Kini U, DDD Study, Hurst JA, Clayton-Smith J, Mehta SG, Toriello HV, Sapp JC, Biesecker LG, Banks S, Basel DG, Sima A, Vance GH, Hickey SE, Alkuraya FS, Abdulrahman O, Fatani E, Hashem M, Bhat MD, Kamate M, Kothari S, Kiran VS, Phadke SR, Kabra M, Puri RD, Kulshreshtra S, Verma IC, Phadke SR, Ghosh M, Radhakrishnan AG, Ramachandran R, Thomas MM, Pizzuti A, Bocchinfuso G, Savasta S, Mattina T, Melis D, Mostardini M, Gurrieri F, Gandini G, Sensi A, Calzolari E, Garavelli L, Brusco GF, De Brasi D, Brunetti-Pierri N, Ferraris S, Ferrero GB, Silengo M, Picco P, Sacco M, Nozza P, Di Rocco M. The spectrum of DNMT3A variants in Tatton-Brown-Rahman syndrome overlaps with that in hematologic malignancies. Am J Med Genet A, 2017.PMID 28941052
- [12]Russo S, Milani D, Meossi C, Finelli P, Selicorni A, Mainini I, Mammi I, Valli M, Colapietro P, Bulfamante G, Rusconi S, Macchi M, Bruschi R, De Santis D, Pedrazzani F, Caporali C, Lapi E, Sala C, Bedeschi MF, Coltrinari D, Giardino D, Tenconi R, Palumbo P, Carella M, Grosso E, Bonfante E, Faleschini E, Vaccari C, Fabbretti M, Calzari L, De Silvestri A, Di Candia S, Bosio L, Macellaro P, Palumbo O, Palumbo P, Carella M, Martinelli P, Cavaliere ML, Grenci A, Lombardo V, Belligni EF, Battista M, Natacci F, Donati F, Pollazzon M, Gangi S, D'Arrigo S, Maitz S, Fittipaldi F, Piscosquito G, Berardo A, Esposito S, Tessadori F, Wit JM, Hennekam RC, Tomà P, Castori M, Silengo MC, Miozzo M, Ferrero GB, Larizza L. Beckwith-Wiedemann spectrum (BWSp): an update on diagnosis, management, and follow-up from the Italian BWSp Register. Ital J Pediatr, 2025.PMID 41126215