Paeds · ent-hearing-and-oral-health
Hearing assessment and childhood hearing loss
Also known as Hearing assessment and childhood hearing loss · Paediatric hearing loss · Newborn hearing screening · Permanent childhood hearing loss · Glue ear and conductive hearing loss
Fellowship guide to hearing assessment and childhood hearing loss: how universal newborn hearing screening (AABR and OAE) and the 1-3-6 milestones catch permanent congenital hearing loss, the conductive-versus-sensorineural framework with otitis media with effusion as the commonest acquired cause, the aetiology (genetic including GJB2, congenital CMV as the commonest infective cause, auditory neuropathy spectrum disorder), age-appropriate audiometry and tympanometry, grommet criteria with three months of watchful waiting and the Paradise developmental evidence, hearing aids and cochlear implantation, and the principle that the open critical period makes early identification non-negotiable.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A baby who does not startle to a hand-clap at six months is the picture most clinicians carry of childhood deafness, yet the whole point of modern paediatric hearing care is that this picture should never be the presenting one. Universal newborn hearing screening finds permanent congenital hearing loss in the first weeks of life, before any parent or clinician could notice it, because the brain's window for learning to process sound and language opens in infancy and narrows with every passing month. Childhood hearing loss is therefore a screening-driven, time-critical diagnosis in which the paediatrician's job is to keep every child moving along a audited pathway, not to wait for symptoms. [1] [6]
Hearing loss in a child is any reduction in the sensitivity or the neural processing of sound that interferes with the detection of speech and the development of spoken language, and it is conventionally separated by where the fault lies. A conductive loss sits in the outer or middle ear, where sound is mechanically impeded before it reaches the cochlea; a sensorineural loss sits in the cochlea or the auditory nerve; and a mixed loss has elements of both. The distinction matters because the causes, the tests and the treatments diverge completely, and because the single commonest acquired childhood hearing loss, otitis media with effusion, is conductive and largely reversible, whereas permanent childhood hearing loss is usually sensorineural and irreversible. [1]
The number that frames the whole topic is roughly one to two babies in every thousand born with permanent hearing loss, with a larger group accumulating by school age as delayed-onset and progressive losses declare themselves. The aim of universal screening, codified as the one-three-six milestones, is to identify these children early enough that the brain still has time to build normal auditory pathways. This topic covers the screening programme, the conductive-versus-sensorineural framework, the aetiology and risk factors, the age-appropriate audiometric tests, the grommet decision for glue ear, and the definitive rehabilitation of permanent loss with hearing aids and cochlear implantation. [2] [1]
Classification
Two axes carry almost all the diagnostic weight in childhood hearing loss: where the lesion sits, and how much it reduces hearing. The site axis divides losses into conductive, sensorineural and mixed, and it maps directly onto the cause and the treatment. Conductive loss localises to the outer or middle ear and is dominated by otitis media with effusion, the commonest acquired cause in young children; sensorineural loss localises to the cochlea or the auditory nerve and is the territory of permanent congenital and childhood hearing loss. Naming the site is the first diagnostic act, and the bone-conduction thresholds on the audiogram settle it. [1] [10]
The degree axis grades how much hearing is lost, read from the pure-tone average across the speech frequencies, and it sets the functional impact and the threshold for intervention. In the scale widely used for children, a loss of 16 to 25 dB is slight, 26 to 40 dB is mild, 41 to 55 dB is moderate, 56 to 70 dB is moderately severe, 71 to 90 dB is severe, and more than 90 dB is profound. Even a mild loss matters in a child, because the quiet consonants that carry meaning in speech sit in the soft end of the hearing range, so a 26 dB loss can blur the difference between similar words and slow language. [1]
A third distinction, by onset and progression, is what makes the newborn screen necessary but not sufficient. Congenital loss is present at birth and is what the screen is designed to catch, but some losses are delayed-onset or progressive, declaring themselves months or years after a passed screen. Congenital cytomegalovirus infection, the enlarged vestibular aqueduct and some GJB2 variants can all cause hearing that is normal at birth and falls later, so a clear newborn screen is a snapshot, not a lifetime guarantee, and any regression in responding or any speech delay must trigger re-assessment. [7] [4]
Epidemiology & Risk Factors
Permanent congenital and early-childhood hearing loss is common enough that every clinician will meet it, but rare enough that the screening programme is what finds it. The systematic review of universal newborn hearing screening programmes puts the prevalence of permanent hearing loss confirmed at the screen at about one per thousand, rising as delayed-onset and progressive losses accumulate through childhood. About half to two-thirds of permanent hearing loss is genetic in origin and the remainder is acquired, and within the genetic group the large majority is non-syndromic, with mutations in the gap-junction protein gene GJB2, or connexin 26, the single commonest cause in many populations. [2] [3]
The acquired causes have a clear hierarchy, and congenital cytomegalovirus infection sits at its top as the commonest non-genetic cause of permanent hearing loss in children. Roughly one in ten infants with congenital cytomegalovirus develops sensorineural hearing loss, the loss can be progressive or late-onset, and across childhood the virus accounts for a substantial fraction of all sensorineural hearing loss. The other major acquired risk factors are the perinatal and neonatal insults captured in the Joint Committee on Infant Hearing risk-indicator list: a stay in the neonatal intensive care unit lasting more than five days, assisted ventilation, extracorporeal membrane oxygenation, hyperbilirubinaemia severe enough for exchange transfusion, and exposure to ototoxic drugs. [7] [4] [8]
On the conductive side, otitis media with effusion is so common in the preschool years that it is best regarded as near-universal rather than abnormal. The peak age is between two and five years, when the Eustachian tube is short and horizontal, and the great majority of effusions resolve spontaneously within three months. The risk factors for persistent glue ear are the same factors that crowd a paediatric clinic: daycare attendance, siblings, passive smoke exposure, craniofacial anomalies such as cleft palate and Down syndrome, and a family history. Identifying which children with glue ear have hearing loss that is persistent and impactful, rather than transient, is the clinical skill. [10] [11]
[4]Pathophysiology
The hearing mechanism converts a pressure wave in air into a nerve signal in the brain, and each step of that conversion can fail, which is why the site of the lesion explains the loss. In the outer and middle ear, sound is gathered and amplified; failure here is a mechanical, conductive problem. In the cochlea, the travelling wave along the basilar membrane deflects the inner and outer hair cells, which transduce vibration into nerve impulses; failure here is sensorineural. In the auditory nerve and brainstem, the synchronised volley of impulses travels centrally; failure here is also sensorineural, and includes the distinct entity of auditory neuropathy. [1]
Otitis media with effusion causes its conductive loss through a simple mechanical principle. Eustachian tube dysfunction fails to ventilate the middle ear, the trapped gas is absorbed, and a sterile effusion accumulates, filling the normally air-filled middle-ear space. The fluid mass-loads the tympanic membrane and the ossicular chain, damping their movement, and the result is a flat, conductive loss, typically in the 20 to 40 dB range, that fluctuates as the effusion comes and goes. Because the cochlea and nerve are intact, sound delivered directly to the skull by bone conduction is heard normally, and this is exactly what the audiogram shows: an air-bone gap. [10]
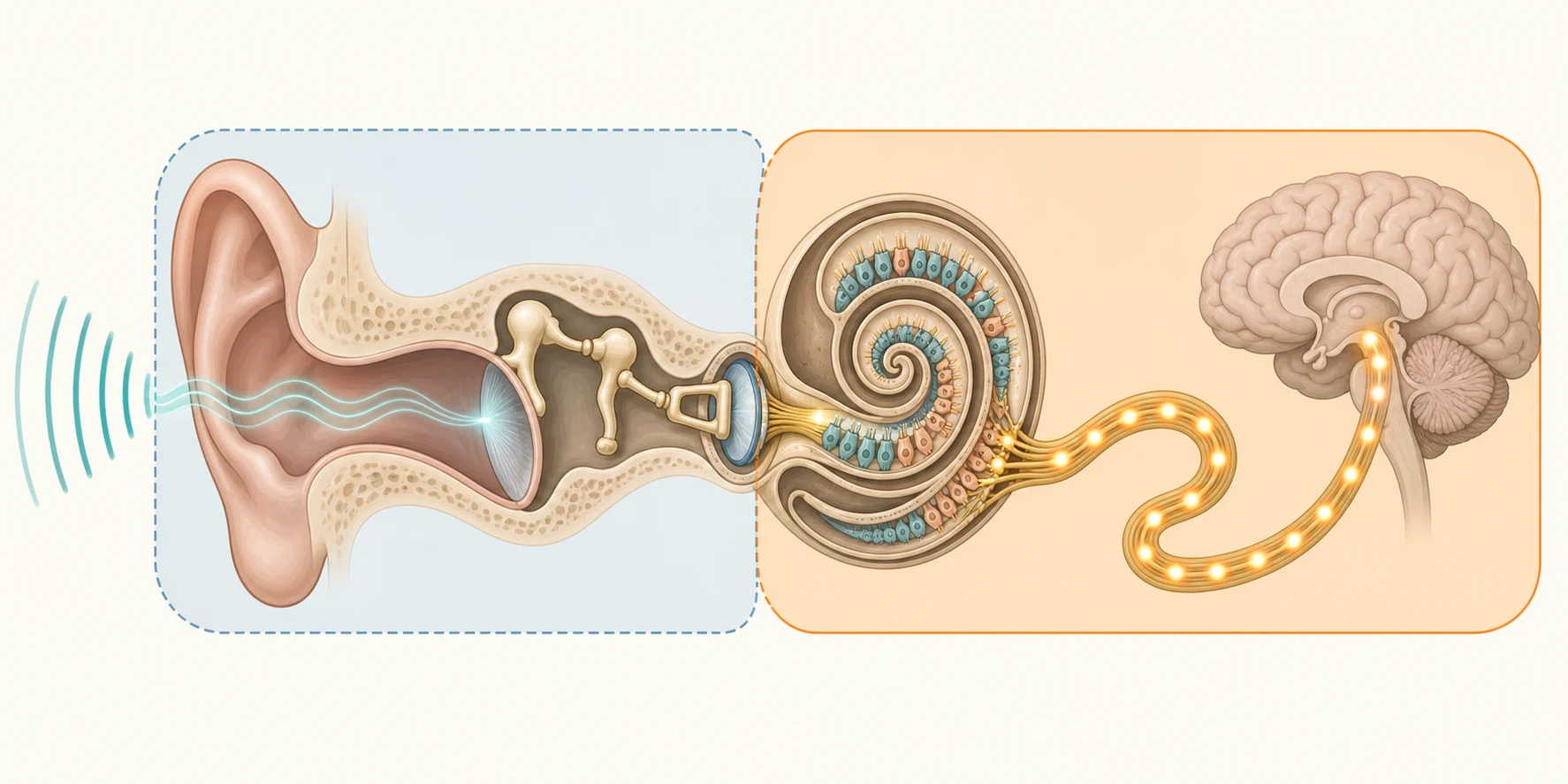
Sensorineural loss is fundamentally a problem of cochlear hair cells or the auditory nerve, and these structures do not regenerate. Genetic causes damage hair-cell function through disrupted cochlear biochemistry; the commonest, GJB2-related connexin 26 deficiency, impairs the recycling of potassium ions in the inner ear, which poisons the hair cells. Cytomegalovirus infects the inner ear directly, producing a cytopathic effect on the hair cells and the stria vascularis. In auditory neuropathy spectrum disorder the outer hair cells survive and the otoacoustic emissions are preserved, but the synchronous nerve discharge that the brain needs to decode sound is lost, which is why an emissions-only screen can read as normal in a child who cannot hear speech. [1] [5] [8]
Clinical Presentation
A newborn with permanent hearing loss is, almost by definition, clinically silent. There is no sign a parent or a bedside clinician can detect, which is precisely why universal screening exists rather than a policy of testing children whose parents are worried. The occasional clue is a risk factor in the history rather than a symptom in the baby: a neonatal intensive care stay, a family history of deafness, features of a syndrome, a known congenital cytomegalovirus infection, or meningitis. The absence of any bedside sign is the strongest argument for screening, and a clinician who reassures a parent that a quiet baby hears is missing the point of the programme. [1] [6]
When hearing loss declares itself after the newborn period, it does so through the child's development and behaviour rather than through a complaint about the ears. The cardinal features are a delay in speech and language, a failure to respond to the name, a child who wants the television loud, who turns to look before localising a voice, or who is withdrawn, inattentive or behaviourally dysregulated at school. Unilateral loss is particularly easy to miss, because the child hears in one ear and converses normally, surfacing only as difficulty hearing in noise or at school. Any of these in a child who passed the newborn screen must prompt re-assessment, never reassurance, because progressive and delayed-onset loss are real. [9] [4]
Otitis media with effusion presents on its own schedule, typically after upper respiratory infections and during the peak preschool years. The child may tug an ear, have mild balance clumsiness, or simply speak more loudly, but the loss is often detected at a routine hearing check or a school entry screen rather than by a specific complaint. The key feature is fluctuation: the hearing comes and goes with the effusion, distinguishing glue ear from the stable, progressive course of a sensorineural loss. A clear history of fluctuating hearing with background upper respiratory infections points to the middle ear. [10]
Differential Diagnosis
The differential begins with the site of the lesion, because that single fork divides the workup. For a conductive loss, the differential is otitis media with effusion, which dwarfs all others, followed by cerumen impaction, tympanic membrane perforation, ossicular fixation or discontinuity, congenital aural atresia or microtia, and cholesteatoma. For a sensorineural loss, the differential is a long list anchored by the genetic causes and congenital cytomegalovirus, with acquired causes such as meningitis, ototoxic drugs, noise and trauma completing it. Tympanometry and the bone-conduction thresholds on the audiogram settle which fork the child is in within minutes. [1] [10]
Within sensorineural loss, the genetic differential divides into non-syndromic, which is the majority, and syndromic, which carries systemic features that may be the clue. GJB2 is the commonest non-syndromic gene. The syndromes an examiner expects are Pendred syndrome, with an enlarged vestibular aqueduct and thyroid disease; Waardenburg syndrome, with pigmentary changes and dystopia canthorum; Usher syndrome, with retinitis pigmentosa; Jervell and Lange-Nielsen syndrome, with a prolonged QT interval that can cause sudden death; and branchio-oto-renal syndrome, with branchial cleft cysts and renal anomalies. A hearing-impaired child is examined for these features, and an electrocardiogram is considered because the cardiac syndrome is lethal if missed. [1] [3]
[1]The must-not-miss acquired causes share the feature that they declare themselves through a clinical event and demand urgent hearing assessment. Bacterial meningitis causes sensorineural loss in a significant fraction of survivors and can trigger rapid cochlear ossification. Congenital cytomegalovirus, confirmed by testing within the first three weeks of life, is the explanation for a child whose loss appears after a normal screen. Ototoxic exposure to aminoglycosides or platinum chemotherapy, and head injury with a temporal bone fracture, complete the short list of events that mandate a hearing test on their own merits, independent of symptoms. [7] [4]
Clinical & Bedside Assessment
The cornerstone of the assessment pathway is the universal newborn hearing screen, delivered before discharge from hospital or within the first month of life. Two technologies carry the screen, and the choice between them turns on a single clinical distinction. Automated auditory brainstem response records the electrical response of the auditory pathway to a click and tests the whole path up to the brainstem, which is why it detects auditory neuropathy. Otoacoustic emissions record the sound that healthy outer hair cells make in response to a stimulus and test only the cochlea, which is fast and cheap but misses any loss that spares the outer hair cells, including auditory neuropathy. [5] [6]
The rule that follows from that distinction is that neonatal intensive care graduates are screened with automated auditory brainstem response, because they carry the highest risk of auditory neuropathy, while well babies may be screened with either. A baby who refers on the screen returns for a repeat, and a baby who refers again is referred for a full diagnostic auditory brainstem response test. The one-three-six milestones govern the whole timetable: screening by one month of age, diagnostic confirmation by three months, and enrolment in early intervention by six months. The paediatrician's role is to audit that every referred baby is moving along this timeline and not being lost. [6]
Beyond infancy, hearing is assessed with the method that suits the child's developmental age, because a pure-tone audiogram needs cooperation that a toddler does not have. Below about six months, behavioural observation audiometry is used; from six months to around two and a half years, visual reinforcement audiometry, in which the child turns toward a sound and is rewarded with a lit toy; from around two and a half to five years, play audiometry, turning the response into a game; and from about four or five years, conventional pure-tone audiometry. The old infant distraction test is unreliable and no longer recommended. [1]
Tympanometry is the bedside test that defines the middle ear and is inseparable from the assessment of the young child with possible conductive loss. A probe in the ear canal changes the pressure and measures the compliance of the tympanic membrane, producing a tympanogram. A type A curve is normal, a type C curve shows negative middle-ear pressure, and a flat type B curve is the hallmark of a middle-ear effusion, the audiometric fingerprint of glue ear. Otoacoustic emissions, quick to perform, screen outer-hair-cell function and are the basis of much newborn screening, while an absent emission combined with a present brainstem response raises auditory neuropathy. [10] [5]
Investigations
The diagnostic gold standard for confirming hearing loss in an infant is the auditory brainstem response, performed with the child asleep or sedated, which gives frequency-specific thresholds without needing cooperation. It distinguishes conductive from sensorineural loss by adding bone conduction, it grades severity, and it confirms or excludes auditory neuropathy when paired with otoacoustic emissions. For the older cooperative child, pure-tone audiometry with both air and bone conduction is the definitive test, and the gap between the two thresholds is the air-bone gap that confirms a conductive component. Speech audiometry then confirms that the loss is sensory and not central. [1] [5]
Once the loss is confirmed and characterised by site and degree, the aetiological workup follows. Imaging is tailored: a computed tomography scan of the temporal bone shows bony malformations such as cochlear dysplasia, semicircular canal anomalies and the enlarged vestibular aqueduct, while magnetic resonance imaging defines the cochlear nerve and is preferred when nerve aplasia or hypoplasia is suspected. Genetic testing, beginning with GJB2 and GJB6 and moving to a gene panel, is offered to every child with unexplained sensorineural loss, because identifying a cause ends the diagnostic odyssey and informs recurrence counselling. [1] [3]
Congenital cytomegalovirus must be sought specifically and early, because the window to confirm a congenital infection is narrow. A polymerase chain reaction test on saliva or urine in the first three weeks of life confirms congenital cytomegalovirus, and the dried blood spot from the newborn screen can be tested retrospectively for a child who presents later, to distinguish congenital infection from an acquired one. The reason to chase this diagnosis is that it is treatable: a six-month course of valganciclovir improves hearing and developmental outcomes in infants with symptomatic congenital cytomegalovirus. An electrocardiogram to exclude the prolonged QT of Jervell and Lange-Nielsen syndrome, and an ophthalmology review, complete the standard workup of a child with unexplained sensorineural loss. [7] [8]
Management — Resuscitation
The word resuscitation sits oddly next to a chronic sensory condition, but childhood hearing loss has a genuine time-critical limb in which delay causes irreversible harm. The clearest example is sensorineural hearing loss after bacterial meningitis, which is an audiological emergency rather than a routine referral. The inflammation can trigger new bone formation, or ossification, within the cochlea within weeks of the infection, which narrows or closes the window for a cochlear implant electrode. Every child who recovers from bacterial meningitis must therefore have hearing assessed urgently before discharge and again in the following weeks, and a child who loses hearing needs early implantation before the cochlea ossifies. [1] [4]
The second time-critical principle applies to every child in the programme, not only the dramatic case: the critical period of auditory development will not wait. A baby who refers on the newborn screen and is lost to follow-up loses months of cortical development that early amplification or implantation would have preserved, and the loss is never fully recovered. The resuscitation, in this sense, is the relentless auditing of the pathway: every refer is tracked, every diagnostic appointment is kept, and no child drifts past six months without enrolment in early intervention. The intervention is not an emergency drug, but the speed of the system is treated with emergency seriousness. [6] [2]
The time-critical actions in childhood hearing loss
Screen every baby by one month of age with AABR for NICU graduates or OAE or AABR for well babies
Track every refer to a diagnostic auditory brainstem response by three months
Assess hearing urgently after bacterial meningitis, before discharge and within weeks, because the cochlea can ossify
Confirm congenital CMV by PCR within the first three weeks if the loss is suspected, and offer valganciclovir for symptomatic infection
Enrol every confirmed child in early intervention by six months to protect the critical period
Re-assess any child with speech delay or regression, even with a passed newborn screen
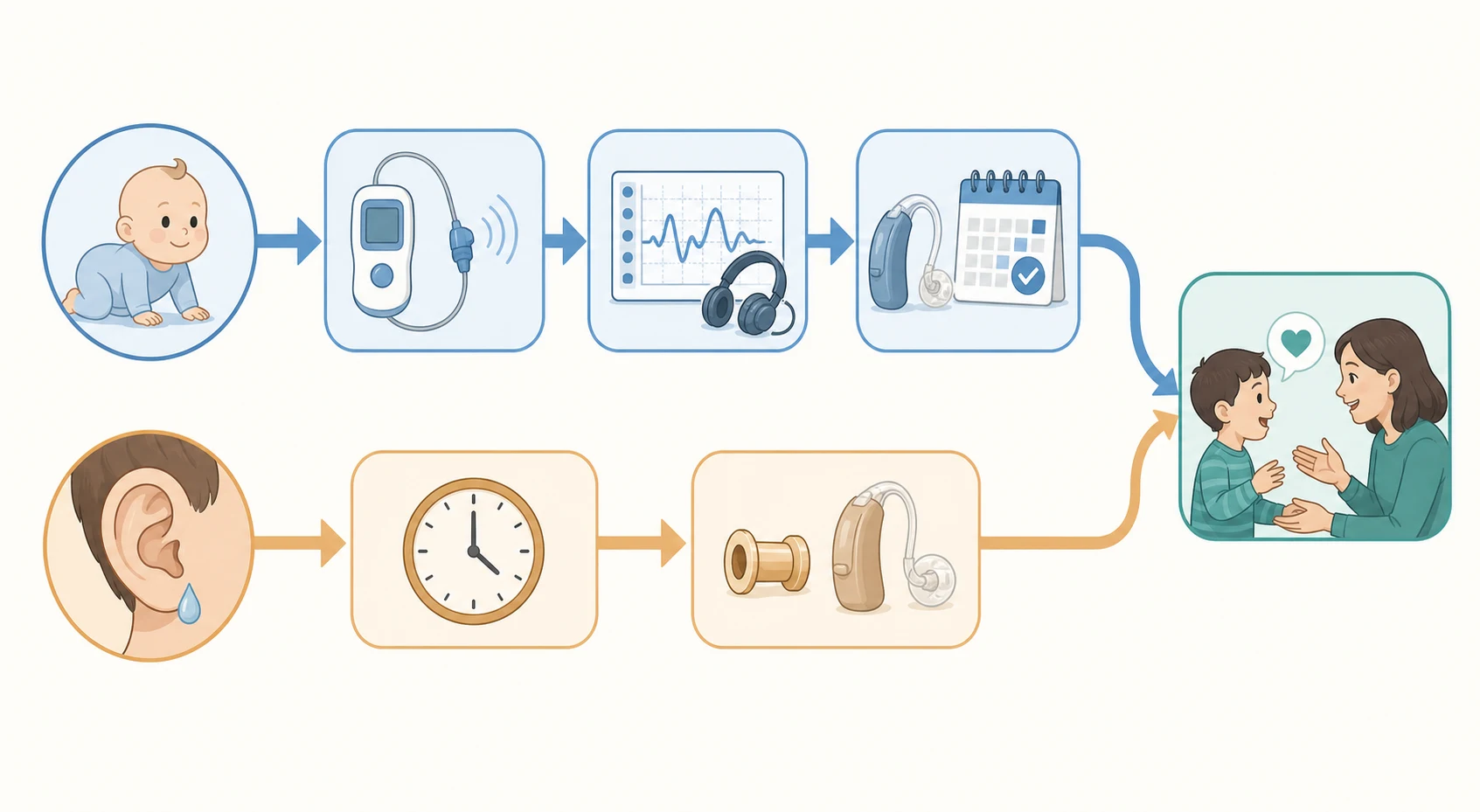
Management — Definitive & Stepwise
The definitive management of a conductive loss caused by otitis media with effusion follows a watch-and-review strategy, because most effusions resolve on their own. The first step is a documented period of watchful waiting for three months from the onset, or from the date of diagnosis if the onset is uncertain, combined with reassurance and a check of the child's development. During this period medical treatments such as antibiotics, antihistamines and steroids have no lasting benefit and are not used. The minority whose effusion persists with a meaningful hearing loss move to surgical management. [10]
The surgical treatment is the insertion of ventilation tubes, or grommets, and the decision is governed by clear criteria rather than by the mere presence of an effusion. The widely accepted threshold is persistent bilateral otitis media with effusion for three months or more with a hearing level of 25 to 30 dB or worse, recorded over the watchful-waiting period, particularly when the loss affects speech, behaviour or education. Children at higher risk of developmental impact from glue ear, including those with Down syndrome, cleft palate and permanent hearing loss in the other ear, have a lower threshold for grommets because even a small additional conductive loss compounds their disadvantage. [10] [11]
Ventilation tubes (grommets) for otitis media with effusion
Loading dose
Not applicable — surgical insertion under brief general anaesthetic
Maintenance dose
Tubes extrude spontaneously over 12 to 18 months; hearing re-checked after insertion and at intervals
The evidence that tempers enthusiasm for grommets is among the most cited in paediatric otolaryngology. The Cochrane review of ventilation tubes for otitis media with effusion confirms a modest improvement in hearing that fades as the tubes extrude, but the randomised trials led by Paradise found that inserting tubes early, rather than waiting, made no difference to developmental, cognitive or academic outcomes at three, six and nine to eleven years of age. Grommets are therefore offered for the hearing and the functional impact they provide over the months they sit in place, not as a developmental safeguard, and watchful waiting remains the default for the majority. [11] [12]
[10]For permanent sensorineural loss, definitive management is rehabilitation, and its form is chosen by the degree and the site of the loss. Mild to severe losses are managed with hearing aids, which amplify sound; for a child with congenital aural atresia in whom sound cannot reach an intact cochlea through the ear canal, a bone-conduction device delivers vibration directly through the skull. Children with auditory neuropathy spectrum disorder may derive limited benefit from conventional amplification and often benefit from frequency-modulated systems and visual support, with cochlear implantation for selected cases. The guiding principle is that the device is fitted early and adjusted as the child grows, in a family-centred programme. [1] [6]
Profound sensorineural hearing loss, in which a hearing aid provides insufficient benefit, is treated with cochlear implantation, which bypasses the damaged hair cells and stimulates the auditory nerve directly. The pre-lingual deaf child gains the greatest advantage from implantation in the first or second year of life, within the critical period, and bilateral implantation is standard where resources allow. Meningitis-related loss is implanted early, before cochlear ossification closes the surgical window. Implantation is followed by intensive auditory-verbal therapy, and outcomes in young, well-supported children are excellent, with many entering mainstream education and acquiring spoken language. [1]
Specific Subtypes & Scenarios
Congenital cytomegalovirus infection is the single scenario an examiner most wants to see handled in full, because it is the commonest non-genetic cause of permanent hearing loss and one of the few with a medical treatment. The hearing loss is sensorineural, often progressive or late-onset, and it can appear in a child whose newborn screen was clear, which is why any unexplained or worsening loss should prompt a search for the virus. Confirmation requires a polymerase chain reaction test on saliva or urine within the first three weeks of life for a congenital diagnosis, or retrospective testing of the newborn dried blood spot. The longitudinal studies show that the loss can emerge and progress over years, so these children need prolonged audiological surveillance. [7] [8] [9]
Otitis media with effusion is the everyday scenario, and the skill is separating the transient majority from the minority who need intervention. The child presents with fluctuating hearing loss in the preschool years, often after colds, the tympanogram is flat and type B, and the audiogram shows a conductive loss with an air-bone gap. Three months of watchful waiting resolves most cases; the child who persists with 25 to 30 dB or worse bilateral loss, or who is in an at-risk group, is offered grommets for the functional benefit, understanding that the developmental evidence does not favour early surgery. The safety-netting is that hearing is rechecked, because effusions recur. [10] [11]
Auditory neuropathy spectrum disorder is the scenario that traps the unwary, because it is invisible to an emissions-only screen. The outer hair cells function, so the otoacoustic emissions are present, but the auditory brainstem response is abnormal or absent, reflecting disordered neural synchrony. It is commonest in neonatal intensive care graduates and in children with hyperbilirubinaemia and prematurity, which is why these babies are screened with automated auditory brainstem response rather than emissions. The child hears tone but struggles to decode speech, and management is individualised, ranging from hearing aids and frequency-modulated systems to cochlear implantation. [5] [1]
In Australia and New Zealand, universal newborn hearing screening is delivered through national programmes, with diagnosis and rehabilitation supported by Australian Hearing and the National Acoustic Laboratories and, in New Zealand, by the national screening programme. A particular regional priority is the disproportionate burden of otitis media and chronic suppurative otitis disease in Aboriginal and Torres Strait Islander and Maori and Pacific children, driven by overcrowding, early daycare exposure and reduced access, which makes aggressive early detection and management of middle-ear disease in these communities a public-health priority rather than an individual concern. Telehealth audiology extends reach into rural and remote areas. [1]
Unilateral hearing loss is the easily missed scenario, affecting roughly three to six per thousand school-age children. The child converses normally and passes a quiet-room screen, but struggles to hear in background noise and to localise sound, which affects learning and fatigue at school. The management is educational support, preferential classroom seating, a frequency-modulated system, and sometimes a contralateral routing of signal aid, rather than reassurance that one good ear is enough, because the long-term impact of an unaided unilateral loss on literacy and behaviour is real. [1]
Complications & Pitfalls
The dominant complication of childhood hearing loss is the developmental one, and it is entirely a function of delay. A child whose permanent hearing loss is identified late, or whose intervention is delayed, accumulates deficits in speech, language, literacy, social interaction and educational attainment that compound through childhood and persist into adult life. The whole architecture of universal screening exists to prevent this complication, and the gap between an identified-at-six-months child and an identified-at-three-years child, given identical technology, is the measure of how much is at stake in keeping the pathway moving. [6] [1]
The complications of the specific causes follow their own courses. A missed congenital cytomegalovirus infection means a child loses the chance for antiviral treatment that could stabilise or improve hearing, and loses the explanation for a progressive loss. Untreated chronic suppurative otitis media in a high-prevalence community can progress to cholesteatoma, mastoiditis and permanent conductive or sensorineural damage. Cochlear ossification after meningitis, if implantation is delayed, can make electrode insertion impossible and condemn a child to lifelong deafness. Each of these is preventable with the right action at the right time. [7] [4]
Prognosis & Disposition
The prognosis of permanent childhood hearing loss, when the diagnosis is made early and intervention is timely, is excellent. A child identified through universal newborn screening, confirmed by three months, fitted with appropriate amplification or implanted in infancy, and supported by family-centred early intervention can achieve spoken language and educational outcomes within the range of hearing peers. The prognosis is driven less by the degree of the biological loss than by the age at intervention and the consistency of support, which is why the system's speed is the single most powerful prognostic factor. [6] [1]
The prognosis of the conductive causes is favourable and self-limiting. Otitis media with effusion resolves spontaneously in the great majority within three months, and grommets give a useful window of improved hearing for the persistent minority over the twelve to eighteen months the tubes sit in place, after which the hearing returns to baseline and any recurrence is re-audited. Congenital cytomegalovirus-related loss is the exception that demands vigilance, because it is often progressive and the child needs prolonged surveillance even when the initial loss is mild. [10] [9]
Disposition follows the cause and the pathway. Every newborn who refers is tracked to diagnostic testing and early intervention, with the paediatrician auditing the timeline. A child with persistent glue ear meeting grommet criteria is referred to otolaryngology, while the at-risk child is referred earlier and at a lower threshold. A child with confirmed permanent sensorineural loss is referred to a multidisciplinary hearing service for amplification or implantation, to genetics for aetiological counselling, and to early intervention for language support. A rural or remote child is linked to telehealth audiology and to a regional hearing service so that distance does not delay the pathway. [1] [6]
Special Populations
The neonatal intensive care graduate is the first special population and the one with the most risk factors concentrated. Prematurity, low birth weight, assisted ventilation, extracorporeal membrane oxygenation, ototoxic drugs, severe hyperbilirubinaemia and prolonged stay all raise the risk of both sensorineural loss and auditory neuropathy, which is why these babies are screened with automated auditory brainstem response rather than emissions and why they carry a risk-indicator flag for re-assessment in infancy. Any infant who has spent more than five days in intensive care is on the risk register. [4] [5]
The child with a craniofacial syndrome or chromosomal condition is the second, because these children carry both a structural and a developmental risk of hearing loss. Down syndrome, with its narrow Eustachian tubes and high rate of glue ear, and cleft palate, with its chronic middle-ear ventilation problem, have a much higher burden of conductive loss and a lower threshold for grommets, because an additional conductive loss compounds an existing developmental risk. These children are audited regularly rather than tested once. [10]
Aboriginal and Torres Strait Islander, Maori and Pacific children, and other Indigenous populations, are the third, carrying a disproportionate burden of otitis media and chronic suppurative otitis disease from the earliest months of life. The drivers are overcrowding, early and intense exposure to respiratory pathogens, reduced access to primary care, and the consequences fall as conductive loss that becomes permanent if chronic disease is neglected. The World Health Organization recognises chronic suppurative otitis disease as a priority in high-prevalence Indigenous communities, where aggressive early detection, medical treatment and surgical intervention for the complications are standard. [1]
The rural and remote child, and the migrant, refugee and disadvantaged child, are the fourth and fifth, because the universal screening programme only works if the follow-up reaches every family. Distance, language, health literacy, transience and socioeconomic disadvantage all erode the follow-up rate and widen the gap between a refer and a confirmed diagnosis, which is why audited recall systems, culturally safe services, interpreter support and telehealth audiology are not optional extras but the mechanisms that make the one-three-six milestones real for every child. [6] [2]
Evidence, Guidelines & Regional Differences
The evidence base for universal newborn hearing screening is what underpins the whole programme. The multicentre studies of newborn auditory brainstem response and otoacoustic emissions established that screening could identify hearing loss in the first weeks of life, the systematic review and meta-analysis quantified the yield at around one per thousand confirmed at the screen, and the population studies of follow-up demonstrated that children identified through screening and managed within the one-three-six milestones achieve better language than those identified later. The screening programme is an evidence-based public-health intervention, not a convenience. [5] [2]
The evidence for managing glue ear is among the most thoroughly studied questions in paediatrics. The Cochrane review of ventilation tubes for otitis media with effusion confirms a modest, time-limited improvement in hearing, and the randomised trials of early versus delayed tympanostomy tube insertion, led by Paradise and colleagues, showed no difference in developmental, language, cognitive or academic outcomes at three, six and nine to eleven years. The clinical practice guideline for otitis media with effusion codifies these findings into the three-month watchful-waiting period and the 25 to 30 dB threshold for surgery. Together they make watchful waiting the default and grommets a functional, not developmental, intervention. [11] [12] [10]
[2] [7]The cytomegalovirus literature has transformed the management of one large slice of sensorineural loss. The systematic review confirms that congenital cytomegalovirus is a major risk factor for childhood hearing loss, the longitudinal studies show that the loss is often progressive or late-onset, and the antiviral treatment trials demonstrate that a six-month course of valganciclovir improves hearing and developmental outcomes in symptomatic congenital infection. This evidence makes the pursuit of a congenital cytomegalovirus diagnosis in an affected infant a treatment decision, not merely an aetiological one. [7] [8] [9]
Across ANZ, the United Kingdom, North America and Europe the core is uniform: universal newborn hearing screening before one month, diagnostic confirmation before three months, and early intervention before six months; grommets for persistent bilateral otitis media with effusion at the 25 to 30 dB threshold; and early cochlear implantation for profound loss. What varies is the delivery architecture, the funding of hearing aids and implants, and the intensity of the response to Indigenous ear disease, which is a specific priority in Australia, New Zealand and among other Indigenous populations. The risk-indicator framework of the Joint Committee on Infant Hearing is the common backbone. [1] [4]
Exam Pearls
Newborn hearing screening and the 1-3-6 milestones — 'HEAR'
References
- [1]Korver AMH; Smith RJH; Van Camp G; Schleiss MR; Bitner-Glindzicz MA; Lustig LR; Usami S; Boudewyns AN Congenital hearing loss. Nat Rev Dis Primers, 2017.PMID 28079113
- [2]Butcher E; Dezateux C; Cortina-Borja M; Knowles RL Prevalence of permanent childhood hearing loss detected at the universal newborn hearing screen: Systematic review and meta-analysis. PLoS One, 2019.PMID 31295316
- [3]Liddle K; Beswick R; Fitzgibbons EJ; Driscoll C Aetiology of permanent childhood hearing loss at a population level. J Paediatr Child Health, 2022.PMID 34546616
- [4]Kraft CT; Malhotra S; Boerst A; Thorne MC Risk indicators for congenital and delayed-onset hearing loss. Otol Neurotol, 2014.PMID 25251299
- [5]Sininger YS; Cone-Wesson B; Folsom RC; Gorga MP; Vohr BR; Widen JE; Eilers E; Norton SJ Identification of neonatal hearing impairment: auditory brain stem responses in the perinatal period. Ear Hear, 2000.PMID 11059700
- [6]Watkin PM; Baldwin M Identifying deafness in early childhood: requirements after the newborn hearing screen. Arch Dis Child, 2011.PMID 21047829
- [7]Vos B; Noll D; Whittingham J; Pigeon M Cytomegalovirus - A risk factor for childhood hearing loss: A systematic review. Ear Hear, 2021.PMID 33928914
- [8]Fowler KB; Boppana SB Congenital cytomegalovirus infection. Semin Perinatol, 2018.PMID 29503048
- [9]Goderis J; Keymeulen A; Smets K; Van Hoecke H; De Leenheer E; Boudewyns A; Desloovere C; Kuhweide R; Dhooge I; Deggouj N Hearing in children with congenital cytomegalovirus infection: results of a longitudinal study. J Pediatr, 2016.PMID 26858192
- [10]Rosenfeld RM; Shin JJ; Schwartz SR; Coggins R; Gagnon L; Hackell JM; Hoelting D; Hunter LL; Kummer AW; Payne SC; Moore DS Clinical Practice Guideline: Otitis Media with Effusion (Update). Otolaryngol Head Neck Surg, 2016.PMID 26832942
- [11]MacKeith S; Mulvaney CA; Galbraith K; Webster KE; Rovers MM; Schilder AGM; Brenner MJ; Russell RT; Blackmore H; Daniel M; Williamson IG; Burton MJ Ventilation tubes (grommets) for otitis media with effusion (OME) in children. Cochrane Database Syst Rev, 2023.PMID 37965944
- [12]Paradise JL; Feldman HM; Campbell TF; Dollaghan CA; Colborn DK; Bernard BS; Rockette HE; Janosky JE; Pitcairn DL; Sabo DL; Kurs-Lasky M; Smith CG Effect of early or delayed insertion of tympanostomy tubes for persistent otitis media on developmental outcomes at the age of three years. N Engl J Med, 2001.PMID 11309632