Paeds · fetal-neonatal-and-perinatal
Congenital anomalies and dysmorphic newborn assessment
Also known as Dysmorphic newborn · Birth defects assessment · Congenital malformation evaluation · Dysmorphology examination · Syndromic newborn · Approach to the child with congenital anomalies
Fellowship-level approach to the newborn with a congenital anomaly or dysmorphic features: recognising major malformations and minor anomalies across every body region, clustering findings into a syndromic pattern, the diagnostic ladder from karyotype to chromosomal microarray to rapid exome/genome sequencing, the four mechanisms of congenital anomalies (chromosomal, single-gene, teratogenic, multifactorial), urgent stabilisation of life-threatening defects, genetics referral and family counselling, and ANZ/UK/US/Canada surveillance and programme differences.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A registrar is called to the postnatal ward to see a baby with an unusual face and a clubfoot. The parents are frightened and the question is not one finding but the whole pattern. This is a defining general-paediatrics task, because the answer changes the child's surveillance, the family's recurrence risk, and the conversation for the rest of their lives. [1]
A congenital anomaly is a structural, functional, or metabolic defect present from birth, whether or not it is obvious at delivery. The umbrella term covers malformations (abnormal formation of tissue), deformations (abnormal shaping of normally formed tissue by mechanical force), disruptions (destruction of normally formed tissue), and dysplasia (abnormal organisation of cells). [1]
The dysmorphic newborn is the baby whose constellation of features — usually a mix of one major anomaly and several minor ones — suggests an underlying syndromic or chromosomal cause. The skill is not naming every syndrome from memory; it is recognising that a pattern exists and organising the work-up that will name it. [2]
Separate major anomalies (those with surgical, cosmetic, or functional consequence, affecting roughly 3 per cent of newborns) from minor anomalies (cosmetic variants present in about 15 per cent). A single minor anomaly is usually nothing; three or more minor anomalies raises the probability of an underlying syndrome sharply, because they cluster when a developmental field is disrupted. [1]
Classification
Sort what you see by body region during the head-to-toe sweep, then re-sort it by mechanism once the baby is stable. Region-based thinking drives the examination; mechanism-based thinking drives the investigation and the counselling. [1]

Major versus minor anomalies
- Surgical, cosmetic, or functional consequence
- ≈3 per cent of newborns
- Examples: cleft palate, cardiac defect, spina bifida, omphalocele
- Drives the immediate management and referral
- Cosmetic variant, no serious functional impact alone
- ≈15 per cent of newborns
- Examples: single palmar crease, cupped ear, clinodactyly
- Meaningful only in clusters of three or more — then they flag a syndrome
The re-sort by mechanism comes later and matters for counselling, because recurrence risk is mechanism-dependent. A chromosomal cause (a one-off error in most cases) carries a low recurrence risk, while an autosomal recessive single-gene cause may carry a 25 per cent recurrence risk in future pregnancies. [1]
Hold the distinction between a deformation and a malformation in mind from the start. A deformation, such as positional talipes from oligohydramnios, often corrects spontaneously or with simple measures. A malformation, such as true clubfoot or a cardiac septal defect, reflects intrinsic abnormal development and needs definitive treatment. [9]
Epidemiology & Risk Factors
Congenital anomalies are common as a group even though each individual syndrome is rare, and they are a leading cause of neonatal and under-five mortality globally. Roughly 1 in 33 infants is born with a major congenital anomaly, which is why every clinician who sees newborns will encounter them. [1]
The risk factors cluster into maternal, fetal, and environmental buckets. Advanced maternal age raises the risk of chromosomal aneuploidies such as Trisomy 21. Maternal diabetes, certain anticonvulsants (valproate, carbamazepine), alcohol, and folate deficiency each raise the risk of specific structural defects. [1]
Family history is the single most informative piece of data the parents bring. Consanguinity raises the risk of autosomal recessive conditions, and a previously affected child or a family history of recurrent pregnancy loss points toward a genetic cause worth investigating actively. [1]
Antenatal factors shape the differential before you even examine. Poorly controlled maternal diabetes is associated with cardiac defects, caudal regression, and sacral agenesis. A history of a congenital infection raises the possibility of a teratogenic pattern of anomalies. Always read the antenatal records, because many major anomalies are now detected before birth. [1]
Pathophysiology
A congenital anomaly arises when one of four mechanisms disturbs development. Knowing the mechanism tells you the inheritance, the recurrence risk, and which test will name it. [1]
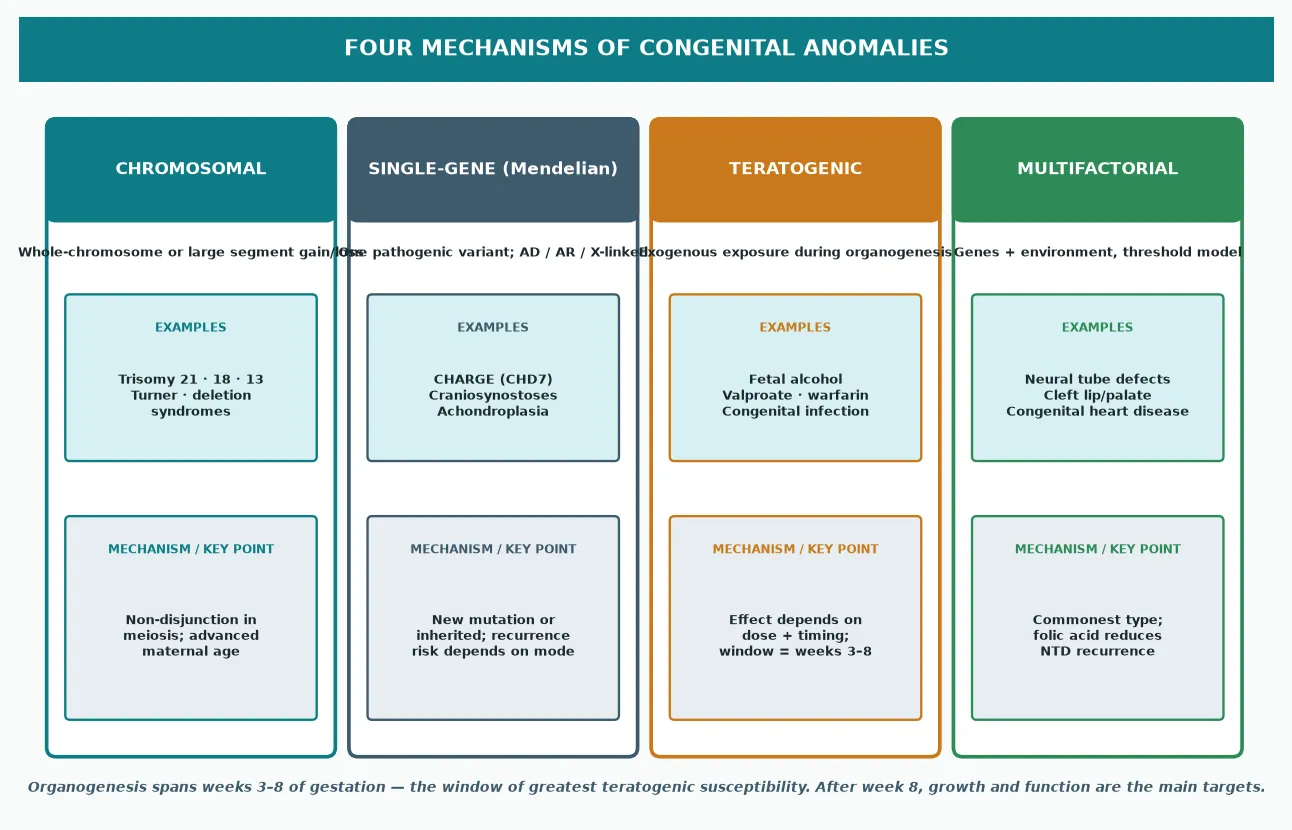
Chromosomal anomalies involve whole chromosomes or large segments and are usually sporadic meiotic errors. Trisomy 21, 18, and 13, and sex chromosome aneuploidies such as Turner syndrome, sit here. Advanced maternal age is the recognised risk factor for the common autosomal trisomies. [3]
Single-gene (Mendelian) disorders follow autosomal dominant, autosomal recessive, or X-linked patterns. Many syndromes once diagnosed only on pattern — CHARGE syndrome, achondroplasia, the craniosynostoses — now have a known gene, which lets you confirm the diagnosis and counsel precisely on recurrence. [10]
Teratogenic anomalies follow an exogenous exposure during organogenesis, and the effect depends on both dose and timing. Alcohol, valproate, warfarin, isotretinoin, and several congenital infections each produce recognisable patterns. Organogenesis spans roughly weeks three to eight of gestation, which is the window of greatest susceptibility. [7]
Multifactorial anomalies reflect a threshold of genetic predisposition plus environment and are the commonest type. Neural tube defects, isolated cleft lip with or without cleft palate, and many congenital heart defects sit here. The clearest demonstration of the mechanism is that preconception folic acid supplementation measurably reduces the recurrence of neural tube defects. [5] [6]
Clinical Presentation
A newborn with a congenital anomaly usually presents in one of three ways: an obvious structural defect at the routine examination, a dysmorphic pattern that an experienced eye notices, or a sick baby whose decompensation reveals an underlying lesion. Each pathway demands a different first move. [1]
The obvious major anomaly announces itself — a cleft lip, an abdominal wall defect, a clubfoot, an open spinal lesion. These are not diagnostic puzzles; they are triage decisions, because the first question is always whether the baby is stable. [4] [9]
The dysmorphic pattern is subtler and is the one that rewards a systematic eye. Look for the craniofacial gestalt — upslanting palpebral fissures and a flat nasal bridge in Down syndrome, low-set posteriorly rotated ears and a flat occiput in Edwards syndrome, the small jaw and midface of Pierre Robin sequence. No single feature makes the diagnosis; the pattern does. [3]
The sick baby with an underlying lesion presents through decompensation. A duct-dependent cardiac lesion, a bowel obstruction from duodenal atresia in Trisomy 21, or adrenal crisis in a virilised infant with congenital adrenal hyperplasia — these are the babies whose anomaly declares itself through physiology rather than appearance. [3]
Minor anomalies are the supporting evidence of a syndromic pattern. Single palmar crease, clinodactyly, sandal-gap toes, Brushfield spots, cupped ears, and a short sternum are individually trivial but, taken together, build the picture that points to a diagnosis. [3]
Growth disturbance often accompanies syndromes. A small-for-gestational-age baby with dysmorphism raises the possibility of a chromosomal cause, a congenital infection, or a skeletal dysplasia. Always plot weight, length, and occipitofrontal circumference, and never accept a single measurement without comparing the three. [3]
Differential Diagnosis
For any single anomaly, hold the isolated form and the syndromic form side by side, because the work-up and the counselling diverge sharply. The discriminator is the rest of the examination. [1]
Isolated versus syndromic presentation
- Isolated VSD / ASD — common, often sporadic
- Syndromic — Trisomy 21 (AVSD), Turner (coarctation), Noonan, 22q11 deletion
- A cardiac anomaly with two minor features → genetics referral and microarray
- Isolated cleft lip ± palate — multifactorial, folate-responsive
- Syndromic — Pierre Robin, Van der Woude, Stickler, 22q11
- Bilateral cleft and a midface defect raise the chance of a syndrome
- Isolated idiopathic — Ponseti method, excellent outcome
- Syndromic — arthrogryposis, myelomeningocele, distal arthrogryposis
- Rigid, bilateral, or accompanied by other anomalies → look harder
- Placental / constitutional — symmetric or asymmetric
- Syndromic — Trisomy 13 / 18, Russell-Silver, congenital infection
- Dysmorphism with growth restriction → chromosomal work-up
The cardinal syndromic associations are high-yield for exams and for practice. Duodenal atresia, atrioventricular septal defect, and a single palmar crease point to Trisomy 21. Clenched hands with overlapping fingers, rocker-bottom feet, and a low posterior hairline point to Trisomy 18. Cleft lip, polydactyly, microcephaly, and holoprosencephaly point to Trisomy 13. [3]
For the dysmorphic newborn with no immediate syndromic match, the differential is deliberately broad and the diagnosis is made by the laboratory, not the eye. Chromosomal microarray, then exome or genome sequencing, resolve a large fraction of these presentations. Hold the expectation that some patterns will remain unexplained even after full testing. [2] [11]
Clinical & Bedside Assessment
Examine warm, with both parents present and an interpreter if needed, and approach the baby calmly. The aim is a complete, documented record of every finding, because a syndrome is a pattern and a pattern is only as good as the notes that capture it. [8]
Observe the gestalt first, before you touch the baby. Step back and let the overall facial impression form — then break it into its parts. Photograph the face in three views (front, profile, and three-quarter), plus hands and feet, because photographs allow later review and are invaluable for the geneticist. [8]
Walk head to toe. Measure the head (occipitofrontal circumference), examine the fontanelles and sutures, the eyes (including red reflex and spacing), ears (position and rotation), mouth and entire palate (run a finger along the posterior palate for a submucous cleft), neck, chest and heart (murmur, femoral pulses), abdomen (masses, wall defects, umbilical vessels), genitalia and anus (patency), spine (midline defects, sacral stigmata), limbs (digits, talipes, reduction), and skin. [1]
A single umbilical artery is a quick, examinable marker — count the vessels in the cord. It is associated with renal and cardiac anomalies in a minority, so it should prompt a renal ultrasound and a careful cardiac examination even when the baby looks well. [1]
VACTERL
VACTERL is a non-random association rather than a single syndrome — at least three component features are required for the label, and there is no single causative gene. Its value is that finding one feature obliges you to hunt for the others, especially the cardiac, renal, and tracheo-oesophageal components that change early management. [1]
Take the family history in three generations, including consanguinity, ethnicity, recurrent miscarriages, stillbirths, and any affected relatives. Draw a pedigree. The family history frequently reframes the recurrence risk and sometimes names the diagnosis before any test. [1]
Investigations
The diagnostic ladder has three rungs, and you climb it in order. Start with the cheapest, most targeted test that the clinical pattern points to, and reserve the powerful broad tests for the unexplained pattern. [2]
Karyotype remains the right first test when the phenotype points clearly to a whole-chromosome aneuploidy such as Trisomy 21, 18, or 13. It is cheap, fast, and answers the targeted question, though it cannot detect the sub-microscopic deletions and duplications that microarray catches. [3]
Chromosomal microarray is the first-tier test for a child with multiple congenital anomalies or developmental delay of unknown cause. It detects copy-number variants — sub-microscopic deletions and duplications — that a karyotype misses, and consensus guidelines established it as the preferred frontline genetic test for this group. [2]
Miller et al. 2010 — consensus statement (Am J Hum Genet)
Population: Individuals with developmental disabilities or congenital anomalies undergoing genetic testing
Key finding
Chromosomal microarray detected a clinically relevant copy-number variant in a higher proportion of patients than a standard karyotype, establishing microarray as a first-tier diagnostic test.
Practice change
For the dysmorphic newborn with multiple anomalies and no clear aneuploidy, microarray is the first-line investigation; karyotype is reserved for cases where a whole-chromosome syndrome is clinically suspected.
Rapid exome or genome sequencing is the escalating rung for the critically ill infant with a suspected monogenic disorder, and it has transformed the diagnostic yield in the neonatal intensive care unit. The randomised NSIGHT1 trial and subsequent studies showed that rapid whole-genome sequencing shortens the time to a molecular diagnosis in sick infants, changing management in a meaningful proportion. [11] [12]
Access to rapid genome sequencing varies widely. Tertiary neonatal networks in Australia, the UK, and parts of the US and Canada now offer rapid sequencing for selected critically ill infants, but availability, funding pathways, and turnaround times differ by region. Confirm your local pathway, because the test is most valuable when it is fast and when a result changes acute management. [11] [12]
Targeted organ screening follows the anomaly. An echocardiogram for any syndrome with a cardiac association, a renal ultrasound for a single umbilical artery or a VACTERL work-up, a hearing screen for syndromes with sensorineural loss, and a detailed eye examination for syndromes with coloboma or cataract. These tests define the full phenotype and shape surveillance. [10]
Management — Resuscitation
A few congenital anomalies are immediate emergencies, and they come before any diagnostic thinking. Recognise them, stabilise, and escalate. [1]
Airway and breathing. Bilateral choanal atresia, severe micrognathia with glossoptosis in Pierre Robin sequence, and large airway malformations can obstruct the neonatal airway. Position, an oral airway, or, in Pierre Robin, prone positioning may relieve obstruction; prepare for definitive airway support and involve the airway team early. [4]
Cardiac. A syndrome with a duct-dependent lesion — hypoplastic left heart in some chromosomal disorders, coarctation in Turner syndrome — may collapse as the duct closes. If saturations are low or differential, start a prostaglandin infusion to keep the duct open and arrange urgent echocardiography and cardiology transfer. [3]
Abdominal wall defects and obstruction. An omphalocele or gastroschisis needs the viscera protected with sterile wrap, heat and fluid loss prevented, and urgent surgical transfer. Bilious vomiting in any newborn is malrotation until proven otherwise, and in Trisomy 21 a duodenal atresia is the classic cause — make the baby nil-by-mouth and involve surgery. [3]
Management — Definitive & Stepwise
Once the baby is stable, the work-up is a sequence of recognise, document, test, refer, and counsel. The diagnosis is built over time, rarely at a single visit. [2]
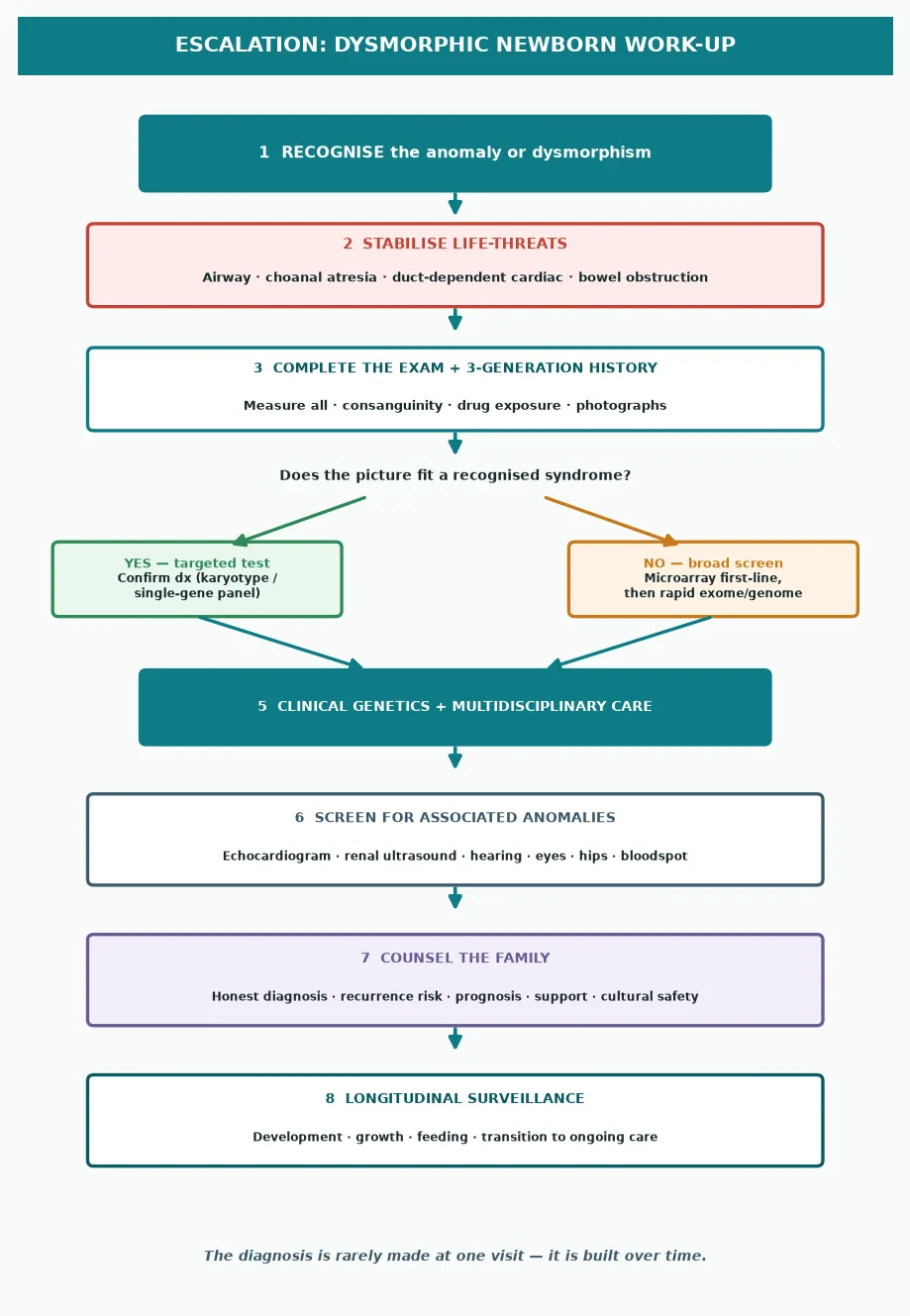
The stepwise work-up of the dysmorphic newborn
Stabilise any life-threatening defect — airway, duct-dependent cardiac, bowel obstruction, adrenal crisis.
Complete the head-to-toe examination, measure all growth parameters, and photograph the findings in three views.
Take a three-generation family history, draw a pedigree, and ask explicitly about consanguinity and drug or teratogen exposure.
Choose the genetic test by the pattern — karyotype for a clear aneuploidy, microarray first-line for multiple anomalies, rapid exome or genome for the unexplained sick infant.
Screen for associated anomalies — echocardiogram, renal ultrasound, hearing, eyes, hips — guided by the suspected diagnosis.
Refer to clinical genetics and the relevant surgical and medical specialties for multidisciplinary management.
Counsel the family honestly and jargon-free — diagnosis, prognosis, recurrence risk, and support services.
Set up longitudinal surveillance — growth, development, feeding, and transition to ongoing care.
The definitive treatment of the anomaly itself follows the organ involved. Clubfoot is corrected with the Ponseti method of serial casting and, for some, a tenotomy, with excellent long-term function. Cleft lip and palate are repaired in a staged surgical sequence with feeding and speech support. Neural tube defects need neurosurgical closure, infection prevention, and long-term bladder, bowel, and mobility management. [9] [5]
The communication is part of the management, not an add-on. Explain the finding in plain language, name the next step and its timing, and give the parents a single trusted clinician to contact. Parents remember how the diagnosis was given for the rest of their lives, and a calm, honest, unhurried conversation is therapeutic in itself. [1]
Specific Subtypes & Scenarios
Each presentation has its own pathway. Walk through the ones a fellowship candidate must defend. [1]
The Down syndrome (Trisomy 21) newborn may show hypotonia, a flat facial profile, upslanting palpebral fissures, a single palmar crease, and a sandal-gap between the first and second toes. The urgent associated findings are an atrioventricular septal defect (echo every baby), duodenal atresia (a "double bubble" on imaging), and later hypothyroidism and hearing loss. Confirm with karyotype and counsel on the recurrence risk. [3]
The Trisomy 18 (Edwards) and Trisomy 13 (Patau) newborns are gravely ill with a recognisable pattern — clenched overlapping fingers and rocker-bottom feet in Edwards; cleft lip, polydactyly, and holoprosencephaly in Patau. Most have major cardiac defects, and the prognosis is poor. Honest, compassionate counselling and shared decision-making about the intensity of care are central. [3]
The cleft lip and palate baby needs feeding support (specialist teats), a staged surgical plan, and screening for an associated syndrome such as Pierre Robin sequence, Stickler syndrome, or 22q11 deletion. Isolated cleft is multifactorial and folate-responsive; syndromic cleft carries the genetics work-up of its syndrome. [4]
The neural tube defect baby — myelomeningocele — needs the lesion protected with sterile saline dressing, neurosurgical referral, antibiotic cover, and screening for associated hydrocephalus and Chiari malformation. Folic acid supplementation before conception prevents a large fraction of neural tube defects, which is why primary prevention sits at the centre of public-health policy. [5] [6]
The clubfoot (talipes equinovarus) baby is managed with the Ponseti method of serial manipulation and casting, with a percutaneous Achilles tenotomy for most, followed by a foot-abduction brace. Idiopathic clubfoot has an excellent long-term outcome when treated early; syndromic clubfoot is more rigid and harder to correct. [9]
The fetal alcohol spectrum disorder pattern — a smooth philtrum, thin vermilion border, and small palpebral fissures, with growth restriction and neurodevelopmental effects — is a teratogenic diagnosis made on the facial features plus a confirmed exposure history. There is no single confirmatory test, so structured criteria and a careful exposure history are essential. [7]
The CHARGE syndrome newborn may present with coloboma, choanal atresia, cranial nerve dysfunction, and characteristic ears. Choanal atresia is the airway emergency that often brings the baby to attention, and CHD7 testing confirms the diagnosis. The constellation demands screening for the cardiac, hearing, and feeding components. [10]
The disorder of sex development presenting as ambiguous genitalia is both an endocrine and a psychosocial emergency. Exclude congenital adrenal hyperplasia by checking electrolytes and 17-hydroxyprogesterone, stabilise if there is salt-wasting, and involve the endocrine and genetics teams before any sex is assigned or communicated. [1]
Complications & Pitfalls
The errors in the dysmorphic newborn are predictable, and naming them is most of the defence. [2]
The classic miss is fixating on one obvious anomaly and missing the pattern. A clubfoot or a cleft is the thing you see; the associated cardiac, renal, or chromosomal finding is the thing that matters. Always complete the head-to-toe examination, even when one finding dominates the picture. [9] [4]
Dismissing minor anomalies as normal variants is the mirror error. A single minor anomaly usually is normal, but three or more is a flag, and clusters build the syndromic picture. Train the eye to record every minor feature rather than mentally editing them away. [3]
Choosing the wrong genetic test wastes time and money. A karyotype for a clearly aneuploid picture, microarray for multiple anomalies of unknown cause, and rapid sequencing for the sick unexplained infant — these are the rungs, and climbing out of order delays the diagnosis. [2] [11]
Missing a duct-dependent cardiac lesion or an adrenal crisis behind an interesting face is the dangerous error. The dysmorphic baby is still a baby, and the airway, breathing, circulation, and glucose come first, always, before the syndromic puzzle. [3]
Poor communication turns a manageable diagnosis into a catastrophe of trust. Vague hedging, breaking the news without both parents present, or offering a recurrence risk you have not verified — each of these harms the family. Honesty, plain language, and a follow-up plan are the standard. [1]
Prognosis & Disposition
The outcome depends almost entirely on the underlying diagnosis and on whether the life-threatening components were caught in time. Most isolated anomalies, treated well, have an excellent prognosis. [1]
Isolated clubfoot treated with the Ponseti method gives a pain-free, functional foot that lasts into adult life. Isolated cleft lip and palate repaired in a staged sequence gives normal speech and appearance with the right support. Isolated cardiac defects repaired in childhood carry a near-normal life expectancy. [9] [4]
The chromosomal trisomies carry a different prognosis. Trisomy 21 has a life expectancy into the sixth decade and beyond with modern cardiac surgery and surveillance. Trisomy 18 and 13 carry high first-year mortality, and the conversation is about compassionate, family-centred care. The diagnosis must drive the prognosis, never the assumption. [3]
The long-term disposition is a medical home and a multidisciplinary team. Growth and developmental surveillance, feeding support, hearing and vision checks, and a clear point of contact for the family — these are what turn a diagnosis into a lived, supported life. Transition to adult care is planned early, not at the last minute. [1]
Special Populations
Several groups need a modified assessment, a different counselling frame, or closer follow-up. [1]
Consanguineous families carry a higher background risk of autosomal recessive conditions, and a dysmorphic baby in this setting should lower the threshold for a broad genetic work-up. Take the pedigree carefully and non-judgementally, and involve genetics early. [1]
Migrant, refugee, and asylum-seeking families may have had limited antenatal care and screening, so anomalies may present for the first time on the postnatal ward. Ensure interpreter access, culturally safe communication, and continuity of follow-up, because a single missed appointment can derail a staged surgical plan. [1]
Indigenous families in Australia and Aotearoa New Zealand carry a higher burden of certain congenital conditions and face real barriers to specialist follow-up across distance. Culturally safe care, partnership with Aboriginal and Māori health services, and attention to the social determinants of access are part of the clinical standard. [1]
Families in rural and remote settings need a clear retrieval and follow-up plan, because the genetics clinic and the surgical centre may be hundreds of kilometres away. Telehealth genetics and supported local teams increasingly bridge this gap. [11]
Socioeconomically disadvantaged families face the same diagnosis with fewer resources, and the practical barriers to follow-up — transport, time off work, cost — are clinical problems, not social footnotes. Build the support around the plan or the plan will fail. [1]
Evidence, Guidelines & Regional Differences
The evidence base for the dysmorphic newborn has shifted in the last decade from pattern recognition toward genomic diagnosis, and the regional programmes differ in how they deliver surveillance and testing. [2]
The microarray consensus established chromosomal microarray as a first-tier test for congenital anomalies and developmental disability, moving it ahead of karyotype for the unexplained pattern. This is the single most influential guideline in modern dysmorphology. [2]
Petrikin et al. 2018 — NSIGHT1 randomised controlled trial (NPJ Genomic Medicine)
Population: Critically ill infants and children with suspected genetic disease in intensive care
Key finding
Rapid whole-genome sequencing increased the proportion of infants receiving an etiologic diagnosis within a clinically useful timeframe compared with standard genetic testing.
Practice change
Rapid genome sequencing is now embedded in many tertiary neonatal networks as a diagnostic tool for the unexplained sick infant, shortening time to diagnosis and changing acute management.
The rapid genome sequencing evidence has matured through the NSIGHT1 trial and subsequent cohort and feasibility studies, which together show a diagnostic yield and a clinical-utility signal strong enough to justify funding in selected critically ill infants. Access, not evidence, is now the limiting factor. [12]
In Australia and Aotearoa New Zealand, congenital anomaly surveillance runs through state-based perinatal data collections and the national birth anomalies registers, and clinical genetics is delivered through tertiary paediatric centres with telehealth outreach. Microarray is the standard first-tier test, and rapid genome sequencing is available through selected programs for critically ill infants. Confirm current local pathways, because programme detail is set at the state or district level. [1]
The controversies cluster around the timing and intensity of intervention in the lethal trisomies, the allocation of rapid sequencing resources, and the ethics of returning incidental genomic findings. Each has a growing evidence base and a strong values dimension, and each is best handled with the family at the centre of the decision. [11] [12]
Exam Pearls
A fellowship candidate should carry a small set of one-liners that examiners reward. [3]
Three or more minor anomalies raises the probability of an underlying syndrome — a single minor anomaly is usually normal. [1]
Down syndrome at the bedside: hypotonia, flat face, upslanting fissures, single palmar crease, sandal-gap toes — and screen every baby for an atrioventricular septal defect with an echo. [3]
Chromosomal microarray is first-tier for multiple congenital anomalies of unknown cause; karyotype is reserved for a clear whole-chromosome aneuploidy. [2]
Bilious vomiting in a Trisomy 21 baby is duodenal atresia until proven otherwise — nil-by-mouth and surgical referral. [3]
Choanal atresia: a baby who is cyanotic at rest and pink when crying; failure to pass a nasogastric tube confirms it; bilateral is an airway emergency and links to CHARGE syndrome. [10]
VACTERL is an association, not a syndrome — at least three components are needed, and finding one obliges you to hunt for the cardiac, renal, and tracheo-oesophageal features. [1]
Folic acid before conception reduces neural tube defect recurrence — primary prevention is part of the answer, not just early surgery. [6]
Pierre Robin sequence: micrognathia, glossoptosis, and cleft palate — the airway obstruction is the emergency, managed with prone positioning or an airway adjunct. [4]
References
- [1]Webber DM Developments in our understanding of the genetic basis of birth defects. Birth Defects Research Part A: Clinical and Molecular Teratology, 2015.PMID 26033863
- [2]Miller DT Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics, 2010.PMID 20466091
- [3]Antonarakis SE Down syndrome. Nature Reviews Disease Primers, 2020.PMID 32029743
- [4]Dixon MJ Cleft lip and palate: understanding genetic and environmental influences. Nature Reviews Genetics, 2011.PMID 21331089
- [5]Copp AJ Spina bifida. Nature Reviews Disease Primers, 2015.PMID 27189655
- [6]Arth A A 2015 global update on folic acid-preventable spina bifida and anencephaly. Birth Defects Research Part A: Clinical and Molecular Teratology, 2016.PMID 27418029
- [7]Hoyme HE Updated Clinical Guidelines for Diagnosing Fetal Alcohol Spectrum Disorders. Pediatrics, 2016.PMID 27464676
- [8]Scheuerle AE Defect evaluation by infant photographs in a multicenter pharmaceutical clinical trial. Birth Defects Research, 2020.PMID 31746564
- [9]Cooper DM Treatment of idiopathic clubfoot. A thirty-year follow-up note. The Journal of Bone and Joint Surgery American Volume, 1995.PMID 7593056
- [10]Lalani SR Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. American Journal of Human Genetics, 2006.PMID 16400610
- [11]Petrikin JE The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill children. NPJ Genomic Medicine, 2018.PMID 29449963
- [12]D'Gama AM Evaluation of the feasibility, diagnostic yield, and clinical utility of rapid genome sequencing in infantile epilepsy (Gene-STEPS): an international, multicentre, pilot cohort study. The Lancet Neurology, 2023.PMID 37596007