Paeds · fetal-neonatal-and-perinatal
Conjugated jaundice and neonatal cholestasis, including biliary atresia
Also known as Neonatal cholestasis · Conjugated hyperbilirubinaemia · Biliary atresia · Neonatal hepatitis · Kasai portoenterostomy · Acholic stools
Fellowship guide to conjugated jaundice and neonatal cholestasis: why a split bilirubin must be checked in any prolonged jaundice, the extrahepatic versus intrahepatic framework, the time-critical exclusion of biliary atresia, the diagnostic panel from ultrasound to cholangiography and biopsy, Kasai portoenterostomy and its determinants, supportive and nutritional care, and long-term outcomes including liver transplantation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Conjugated jaundice in a neonate or young infant reflects impaired bile flow, and unlike unconjugated physiological jaundice it is always pathological and never benign. The underlying process is neonatal cholestasis, defined biochemically as a conjugated (direct) bilirubin fraction above 17 micromol per litre or more than 20 per cent of the total bilirubin. Any infant who is still jaundiced beyond two weeks of life if term, or three weeks if preterm, must have a split bilirubin measured rather than being reassured that the colour will fade. [1]
The most time-critical cause is biliary atresia, an obliterative fibro-inflammatory disease of the extrahepatic bile ducts that progresses to biliary cirrhosis unless bile drainage is restored surgically. A Kasai portoenterostomy, performed as early as possible, is the only chance of native-liver survival, and every additional week of delay measurably worsens its chance of success. Because of this, the entire diagnostic pathway for a cholestatic infant is engineered around one question: does this child have biliary atresia, and how quickly can the surgeon be involved. [3]
Cholestasis also encompasses a wide range of intrahepatic and parenchymal disorders, collectively grouped as neonatal hepatitis, including infective, metabolic, genetic, and endocrine causes. These infants need the same urgency of workup even when biliary atresia is ultimately excluded, because metabolic and infective diseases have their own specific, time-sensitive treatments, and all cholestatic infants share the same risks of malnutrition, fat-soluble vitamin deficiency, and progressive liver injury. [1]
Classification
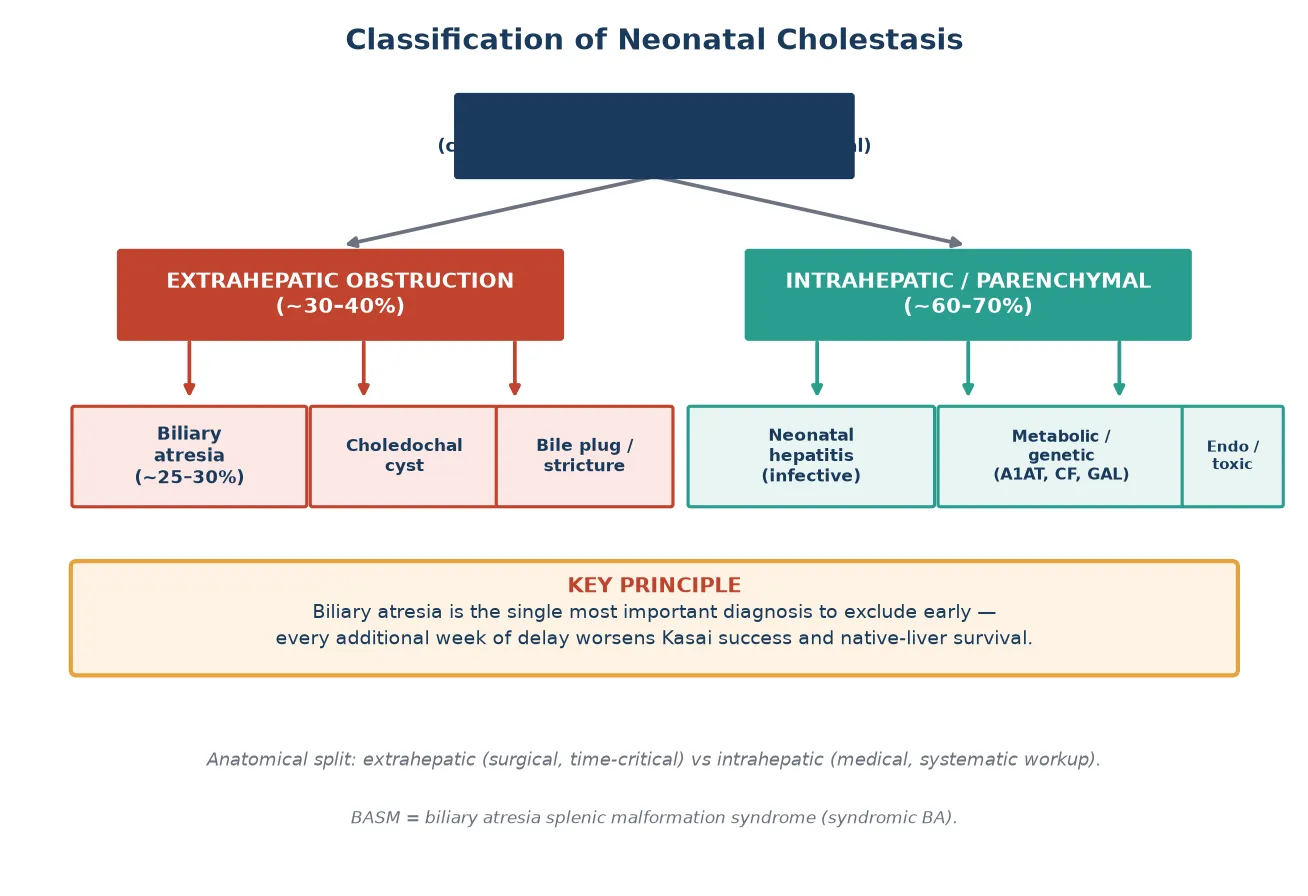
Clinicians classify neonatal cholestasis anatomically into extrahepatic obstruction, where a physical barrier blocks bile flow downstream of the liver, and intrahepatic or parenchymal disease, where the hepatocyte or the intrahepatic bile ducts themselves are dysfunctional. This split matters because the extrahepatic group is surgical and time-critical, while the intrahepatic group demands a systematic medical workup for infection, metabolic disease, and genetic disorders. [1]
[1]Biliary atresia itself is further divided into a perinatal or isolated form, which is by far the most common and presents after a jaundice-free interval, and a syndromic or embryonic form known as biliary atresia splenic malformation (BASM) syndrome. BASM associates biliary atresia with situs abnormalities, polysplenia or asplenia, interrupted inferior vena cava, pre-duodenal portal vein, and cardiac defects, and it carries a worse prognosis than the isolated form. Recognising BASM matters because these infants need echocardiography and a careful search for associated malformations before surgery. [3]
Epidemiology & Risk Factors
Biliary atresia is the single most common indication for liver transplantation in children worldwide, and its incidence varies strikingly by geography and ethnicity. It affects approximately 1 in 8000 to 1 in 18,000 live births in Western populations, while the highest reported rates are in east Asia, particularly Taiwan and Japan, where universal stool-colour-card screening has been adopted specifically because of the burden of disease. [7]
The natural history of untreated biliary atresia is one of relentless progression: without a successful Kasai operation, infants develop biliary cirrhosis, portal hypertension, and end-stage liver failure, and most die within the first two years of life. The single most powerful modifiable determinant of outcome is the age at which the Kasai portoenterostomy is performed, with outcomes deteriorating as surgery is delayed beyond the first 60 to 80 days of life. [4]
For neonatal cholestasis more broadly, risk factors depend on the underlying cause. Infection-related cholestasis is associated with maternal TORCH infections, particularly cytomegalovirus, and with bacterial sepsis. Metabolic cholestasis is suggested by consanguinity, a family history of unexplained infant deaths, and galactosaemia in galactose-exposed infants. Preterm and very low birth weight infants are more prone to cholestasis from prolonged parenteral nutrition, a condition termed intestinal failure-associated liver disease. [1]
Pathophysiology
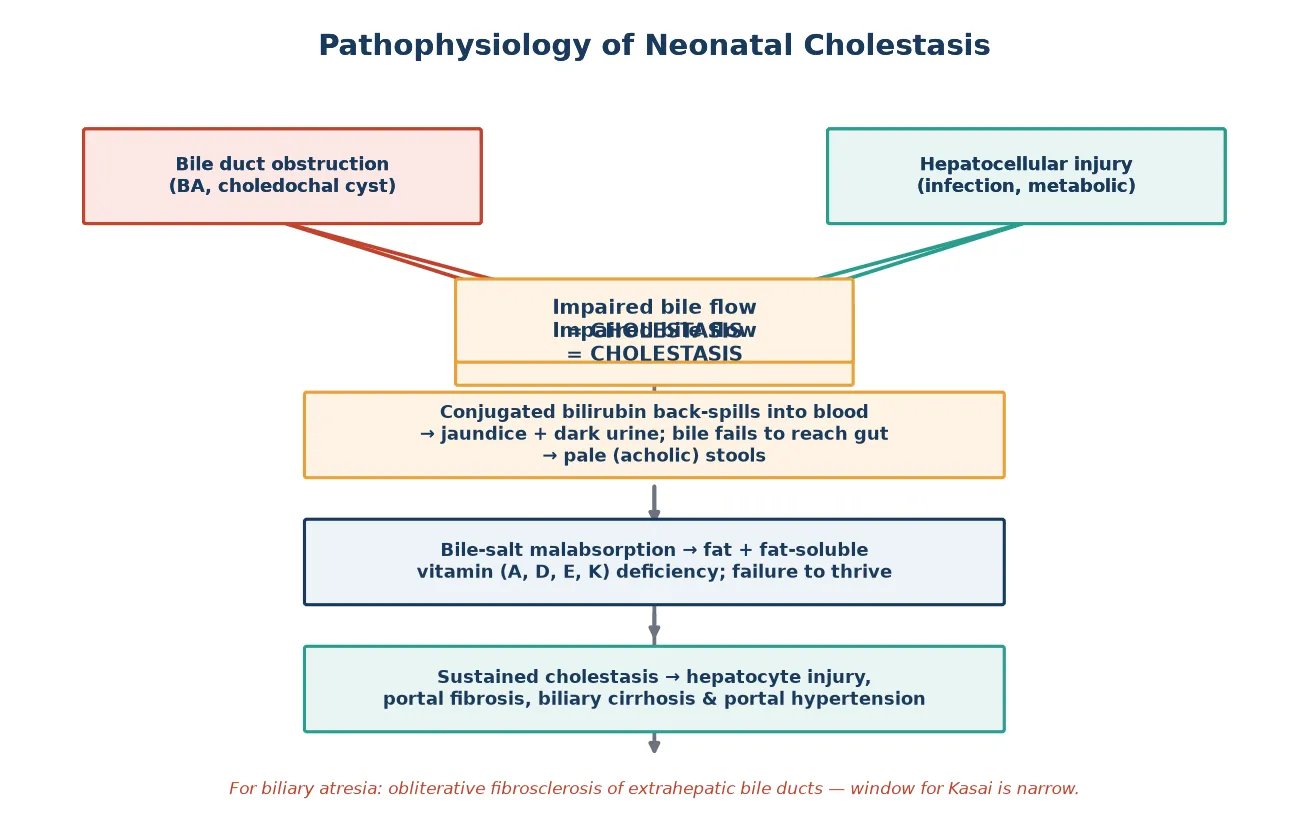
Bilirubin metabolism explains why conjugated jaundice behaves so differently from the unconjugated form. Unconjugated bilirubin, produced from haem breakdown, circulates bound to albumin and cannot cross the renal filtration barrier, so it never appears in urine. Once the hepatocyte conjugates bilirubin with glucuronic acid it becomes water-soluble and is normally excreted into bile. When bile flow is impaired, conjugated bilirubin back-spills into blood and, being water-soluble, is filtered by the kidney, producing the dark, yellow-brown urine that is a hallmark of cholestasis. [1]
In biliary atresia the pathological process is a progressive obliterative fibrosclerosis of the extrahepatic bile ducts. The ducts are normal or dilated in early fetal life but undergo inflammatory destruction after birth, leaving a fibrotic cord at the porta hepatis. The result is complete obstruction of bile outflow: bile cannot reach the gut, so stools lose their bile pigment and become pale or acholic, while bile pressure builds within the liver and drives hepatocyte injury and portal fibrosis. The exact trigger remains uncertain, with proposed roles for viral infection, dysregulated autoimmunity, and developmental signalling defects. [3]
The consequences of sustained cholestasis are shared across all causes and drive much of the supportive care. Failure of bile-salt delivery to the intestine causes fat malabsorption and deficiency of the fat-soluble vitamins A, D, E, and K, leading to failure to thrive, rickets, neuropathy, and coagulopathy. Retained bile salts are pruritogenic, causing distressing itch. Over weeks to months, sustained cholestasis progresses to hepatocyte dropout, bridging fibrosis, biliary cirrhosis, and portal hypertension with splenomegaly, ascites, and variceal bleeding. [1]
Clinical Presentation
The presentation of neonatal cholestasis ranges from an incidental finding of prolonged jaundice in an otherwise well infant to a sick, pale, failing-to-thrive baby with liver failure. Persistent yellow discolouration of the skin and sclera beyond two weeks in a term infant, or three weeks in a preterm infant, is the trigger that demands a split bilirubin rather than further reassurance. The clinician must always ask about and inspect the stool and urine colour directly. [1]
The pathognomonic features of biliary atresia are pale or acholic stools, dark yellow or brown urine that stains the nappy, and progressive jaundice developing over the first weeks of life in an infant who often initially appeared well. Hepatomegaly is usually present, and the liver edge is typically firm. As obstruction persists, splenomegaly develops as a sign of portal hypertension, and the infant may show irritability, poor weight gain, and increasing pallor from the anaemia of chronic disease. [3]
Intrahepatic causes produce a more varied picture that reflects the underlying disorder. Infective neonatal hepatitis from cytomegalovirus, toxoplasmosis, rubella, herpes, or syphilis may present with petechiae, microcephaly, cataracts, or sensorineural deafness. Metabolic disease produces distinctive patterns: galactosaemia causes Escherichia coli sepsis and cataracts, alpha-1-antitrypsin deficiency causes a hepatitis-like illness, and tyrosinaemia causes a cabbage-like odour. Endocrine causes such as hypothyroidism present with hypotonia, a large fontanelle, and prolonged jaundice. [2]
Presenting features to elicit and observe
Jaundice beyond two weeks (term) or three weeks (preterm)
Pale, putty-coloured or acholic stools
Dark yellow-brown urine staining the nappy
Hepatomegaly with a firm edge; later splenomegaly
Failure to thrive, irritability, easy bruising or bleeding
Dysmorphism, cataracts, or cardiac murmur suggesting a syndrome
Differential Diagnosis
The differential diagnosis of neonatal cholestasis is one of the broadest in paediatrics, spanning surgical, infective, metabolic, genetic, endocrine, and toxic causes. The disciplined approach is to work through each category systematically while never losing sight of the surgical priority of excluding biliary atresia. A delay to diagnose a metabolic or infective cause can be as harmful as missing biliary atresia, because many of these disorders have specific, time-sensitive treatments. [1]
The leading extrahepatic causes are biliary atresia, choledochal cyst, bile-plug syndrome, and distal bile-duct stricture. Among the intrahepatic causes, neonatal hepatitis syndrome from infection is common, followed by the metabolic and genetic diseases: alpha-1-antitrypsin deficiency, galactosaemia, hereditary fructose intolerance, tyrosinaemia type 1, cystic fibrosis, bile-acid synthesis defects, and progressive familial intrahepatic cholestasis. Alagille syndrome combines cholestasis with a murmur from peripheral pulmonary stenosis, butterfly vertebrae, posterior embryotoxon, and a characteristic facies. [2]
Endocrine disease, particularly congenital hypothyroidism and hypopituitarism, must always be excluded because the treatment is simple and the consequences of missing it are severe. Toxic causes include prolonged parenteral nutrition, especially in preterm infants, and drug-induced cholestasis. Sepsis itself causes cholestasis, and any unwell cholestatic infant needs a thorough septic evaluation. A neonate with cholestasis and a bleeding disorder warrants urgent consideration of vitamin K deficiency bleeding and coagulopathy. [1]
Clinical & Bedside Assessment
Begin every assessment by inspecting the infant fully undressed in good light. Note the depth and distribution of jaundice, look for dysmorphism, cataracts, and any rash or petechiae, and then move to the abdomen. The clinician must personally look at a fresh stool sample and the wet nappy, because parental report of stool colour is unreliable and acholic stools are frequently missed or misattributed. [1]
Abdominal examination focuses on the liver and spleen. Measure the liver span by percussion and palpation, because a normal liver edge in a neonate may be felt 1 to 2 centimetres below the costal margin but a span above 6 centimetres in a term infant suggests hepatomegaly. The liver edge in cholestasis is typically firm rather than soft. Splenomegaly is a later sign that signals portal hypertension and argues against early biliary atresia. Look for ascites, caput medusae, and digital clubbing as signs of chronic liver disease. [1]
Take a focused history covering the pregnancy, delivery, and feeding. Ask about maternal infections and serology, gestational age, birth weight, feeding type, stool and urine colour changes over time, the age at which jaundice appeared, and any family history of liver disease, consanguinity, or unexplained infant deaths. Document growth, because weight faltering with cholestasis accelerates the need for investigation. Review the newborn bloodspot screening result, because it captures galactosaemia, hypothyroidism, and cystic fibrosis. [1]
Investigations
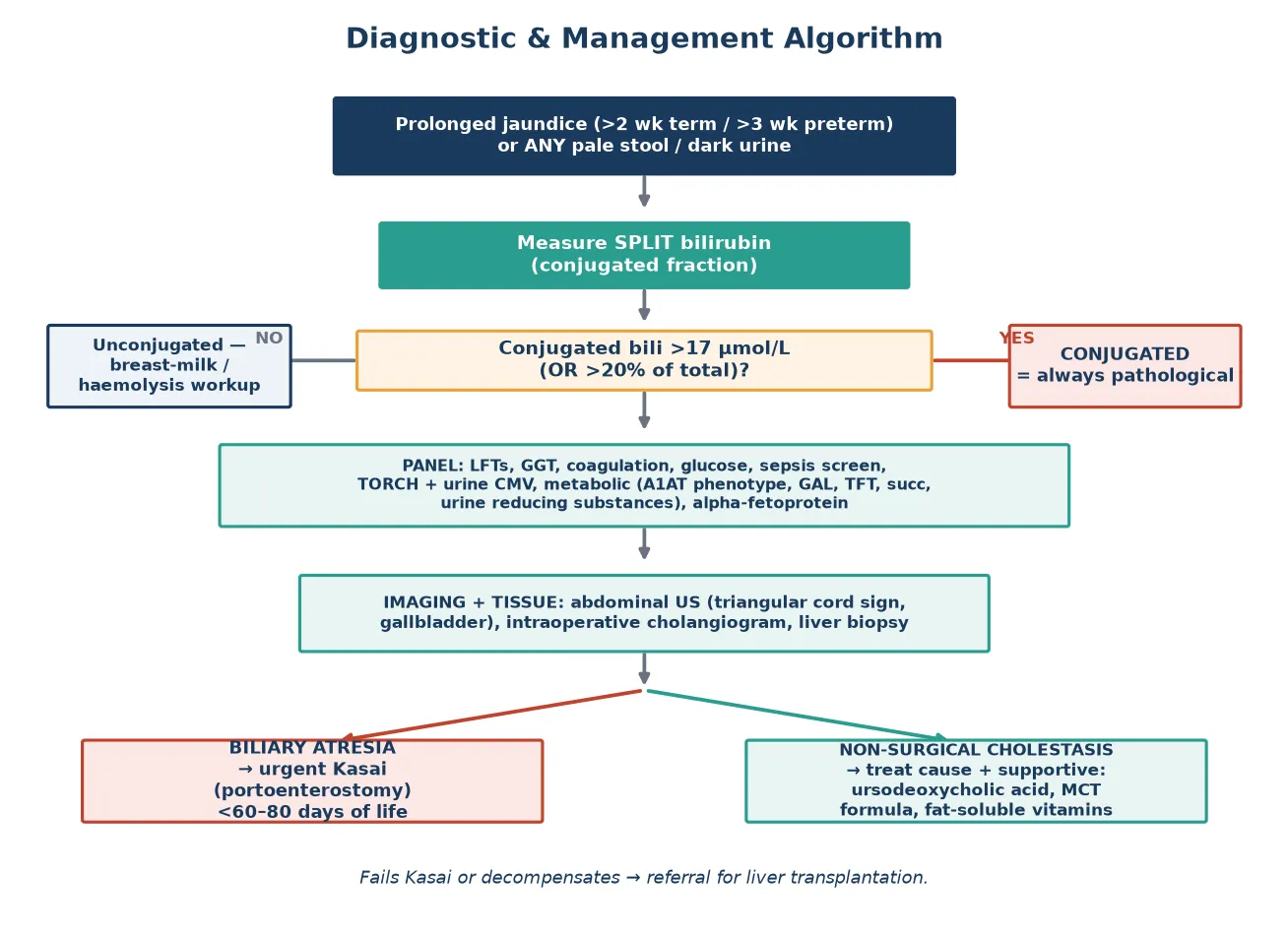
The cornerstone investigation is the split bilirubin, measuring total and conjugated fractions, which must be sent for any prolonged jaundice. A conjugated fraction above 17 micromol per litre or more than 20 per cent of total confirms cholestasis and triggers the full diagnostic panel. Alongside the bilirubin, send a full liver panel including alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, gamma-glutamyl transferase, albumin, and coagulation studies. A high gamma-glutamyl transferase with cholestasis points toward biliary obstruction or biliary atresia, while a low or normal value suggests an intrahepatic cause such as progressive familial intrahepatic cholestasis. [1]
The metabolic and infective workup runs in parallel. Check glucose for hypoglycaemia, send a blood gas and lactate, and measure ammonia if metabolic disease is plausible. Send a full septic screen, TORCH and urine cytomegalovirus polymerase chain reaction, alpha-1-antitrypsin phenotype and level, galactosaemia and tyrosinaemia markers, thyroid function, and urine reducing substances. A sweat chloride test or cystic fibrosis genetic testing excludes cystic fibrosis. Check alpha-fetoprotein, which is markedly elevated in tyrosinaemia. [2]
Imaging begins with an abdominal ultrasound performed after a fast to maximise gallbladder visualisation. In biliary atresia the gallbladder is small or absent and the triangular cord sign, a fibrotic echogenic remnant at the porta hepatis, may be seen, but ultrasound alone cannot exclude biliary atresia. The definitive diagnostic test is an intraoperative cholangiogram performed under anaesthesia, which demonstrates an absent or atretic extrahepatic biliary tree. A percutaneous liver biopsy is highly informative and shows bile-duct proliferation, portal fibrosis, and bile plugs in biliary atresia, distinguishing it from the paucity or hepatitis pattern of intrahepatic disease. [1]
Management — Resuscitation
The initial priority in a cholestatic infant is to identify and treat immediately life-threatening complications. The most dangerous is coagulopathy from vitamin K deficiency, which can cause catastrophic intracranial bleeding. Check the coagulation profile urgently in every cholestatic infant, and if the international normalised ratio is prolonged, give intravenous vitamin K 1 milligram, correcting any deficit before invasive procedures such as biopsy or cholangiography. [1]
Hypoglycaemia is common in metabolic liver disease and in sick infants, so check a bedside glucose and treat with an intravenous dextrose bolus of 2 to 5 millilitres per kilogram of 10 per cent dextrose followed by a maintenance infusion. Sepsis frequently coexists with or precipitates cholestasis, so an unwell infant needs a full septic evaluation and broad-spectrum antibiotics. Establish intravenous access and ensure the infant is well hydrated before any surgical or radiological procedure. [1]
Correct fluid and electrolyte disturbances, particularly in infants with diarrhoea, vomiting, or poor intake. Give parenteral vitamin K 1 milligram and ensure the airway, breathing, and circulation are stable before transfer to a specialist paediatric hepatology and surgical centre. Early liaison with the regional liver unit is essential, because the timing of the Kasai operation depends on rapid referral and transfer, not on completing every non-urgent test first. [1]
Resuscitation priorities in the cholestatic infant
Check coagulation and give intravenous vitamin K 1 mg if INR prolonged
Bedside glucose; treat hypoglycaemia with 2-5 mL/kg of 10% dextrose
Septic evaluation and empiric antibiotics if unwell
Establish IV access and correct dehydration
Stabilise airway, breathing, circulation before transfer
Urgent referral to the regional paediatric hepatology and surgical centre
Management — Definitive & Stepwise
Once biliary atresia is confirmed at cholangiography, the definitive treatment is a Kasai portoenterostomy performed at the same anaesthetic. The surgeon excises the fibrotic extrahepatic bile-duct remnant at the porta hepatis and anastomoses a Roux-en-Y jejunal loop to the denuded liver surface, allowing bile to drain from the intrahepatic ducts directly into the gut. The operation must be performed as early as possible, ideally before 60 days of life, because native-liver survival falls steadily as the age at surgery increases. [4]
Postoperative medical management aims to maximise bile drainage and prevent complications. Ursodeoxycholic acid at 10 to 20 milligrams per kilogram per day in divided doses is widely used to improve bile flow, although robust evidence for survival benefit is limited. Antibiotic prophylaxis against ascending cholangitis, typically oral trimethoprim-sulfamethoxazole, is continued for months because cholangitis attacks threaten native-liver survival. Corticosteroids have been studied in the START trial, which found no significant benefit of high-dose postoperative corticosteroids on the primary outcome of native-liver survival at two years, so their routine use is not evidence-supported. [6]
For all cholestatic infants, regardless of cause, nutritional support is central to management because fat malabsorption causes failure to thrive. Use a medium-chain triglyceride-containing formula, which is absorbed without bile salts, and supplement the fat-soluble vitamins A, D, E, and K parenterally or in water-miscible form. Monitor growth, prothrombin time, and vitamin levels regularly. Ursodeoxycholic acid 10 to 20 milligrams per kilogram per day improves bile flow and pruritus in intrahepatic cholestasis. [1]
VITAMINS for cholestatic supportive care
Liver transplantation is the salvage therapy for infants in whom the Kasai fails, defined as absent bile drainage, recurrent cholangitis, or progression to end-stage liver disease, and for those who present too late for a meaningful Kasai. Biliary atresia remains the leading indication for paediatric liver transplantation globally, and around 70 to 80 per cent of biliary atresia patients will ultimately require transplantation. Excellent long-term graft and patient survival are now achievable. [5]
Specific Subtypes & Scenarios
Biliary atresia splenic malformation syndrome accounts for 10 to 20 per cent of cases and combines biliary atresia with laterality defects. These infants present with jaundice from birth rather than after a jaundice-free interval, and they have polysplenia, asplenia, situs inversus, interrupted inferior vena cava, pre-duodenal portal vein, and cardiac malformations. The Kasai operation is still performed, but native-liver survival is worse than in the isolated perinatal form, and transplant is needed earlier and more often. [3]
Choledochal cyst is a congenital dilatation of the biliary tree that presents with cholestasis, a palpable abdominal mass, or pancreatitis, and is readily identified on ultrasound showing a cystic lesion at the porta hepatis communicating with the bile duct. Treatment is surgical excision of the cyst with a Roux-en-Y hepaticojejunostomy to prevent recurrent cholangitis and the long-term risk of cholangiocarcinoma. Unlike biliary atresia, the urgency is less about days and more about definitive resection. [1]
Parenteral nutrition-associated liver disease, now termed intestinal failure-associated liver disease, is the dominant cause of cholestasis in preterm and very low birth weight infants on prolonged parenteral nutrition. Management focuses on enteral feeding advancement, lipid modification using fish-oil or mixed-lipid emulsions to reduce the hepatotoxicity of soybean-based lipid, and aggressive treatment of sepsis. Most infants recover as enteral feeding is established. [1]
[8]Complications & Pitfalls
The dominant complications of neonatal cholestasis flow from sustained bile-flow failure and progressive liver injury. Ascending cholangitis is a constant threat after a Kasai operation, presenting with fever, worsening jaundice, and acholic stools, and each attack erodes native-liver survival. Portal hypertension develops as fibrosis progresses, bringing splenomegaly, hypersplenism, ascites, and variceal bleeding. Fat-soluble vitamin deficiency causes rickets, neuropathy, night blindness, and potentially fatal coagulopathy from vitamin K deficiency. [9]
Long-term survivors with their native livers face ongoing risks even years after a successful Kasai. Approximately 30 to 50 per cent of biliary atresia patients retain their native liver at 10 years, but most will develop portal hypertension and a small number are at risk of hepatocellular carcinoma, mandating lifelong surveillance with ultrasound and alpha-fetoprotein monitoring. Growth and quality of life are affected by pruritus, chronic disease, and the burden of medications and admissions. [9]
The most dangerous pitfall is reassuring parents that prolonged jaundice is physiological or breast-milk related without measuring a conjugated bilirubin fraction. Other major errors include accepting a normal ultrasound as excluding biliary atresia, delaying referral to complete non-essential tests, failing to give vitamin K before procedures in a coagulopathic infant, missing the metabolic and endocrine causes with specific treatments, and attributing failure to thrive to feeding problems rather than to the underlying liver disease. [1]
Prognosis & Disposition
The prognosis of neonatal cholestasis is determined almost entirely by the underlying cause and, for biliary atresia, by the age at Kasai operation. Infants whose Kasai is performed before 30 to 40 days of life have the best native-liver survival, and outcome declines steadily with each additional week, so that surgery after 90 days carries a poor prognosis for native-liver survival. Centres with high caseloads and standardised centralised care achieve better outcomes, supporting referral to specialist units. [5]
For non-surgical causes, prognosis reflects the specific disorder and its response to treatment. Galactosaemia and hereditary fructose intolerance respond to dietary restriction, congenital hypothyroidism responds to thyroxine, and infective causes may resolve with treatment or time. Progressive familial intrahepatic cholestasis and Alagille syndrome are chronic and often eventually require transplantation. The unifying principle is that all cholestatic infants need structured multidisciplinary follow-up. [1]
Disposition after diagnosis and initial management involves transfer to, or shared care with, a paediatric hepatology centre. After a Kasai operation, infants are monitored closely for bile drainage, cholangitis, and growth, with serial bilirubin, liver function, and coagulation testing. Families need clear education about fever as a red flag for cholangitis, the importance of adherence to antibiotics and vitamins, and the warning signs of variceal bleeding and encephalopathy. A plan for transplantation assessment is established for infants in whom the Kasai fails. [5]
Special Populations
Preterm and very low birth weight infants are a high-risk group for cholestasis, predominantly from intestinal failure-associated liver disease related to prolonged parenteral nutrition, immaturity of bile flow, and recurrent sepsis. Management prioritises early and sustained enteral feeding, minimisation of parenteral nutrition duration, use of fish-oil or mixed-lipid emulsions, and aggressive sepsis treatment. Most preterm infants recover their liver function as enteral nutrition is established, but a minority progress to chronic cholestasis. [1]
Infants in remote and Indigenous communities face barriers of distance, delayed presentation, and reduced access to specialist care that worsen the age at diagnosis of biliary atresia. Stool-colour card screening programmes, culturally safe health communication, and clear referral pathways that do not require multiple gatekeeping steps are the interventions most likely to close this gap. Telehealth links to specialist liver units support remote clinicians in recognising the red flags. [8]
In low- and middle-income settings, late presentation, limited access to paediatric surgery and transplantation, and shortages of medium-chain triglyceride formulas and fat-soluble vitamin supplementation dramatically worsen outcomes for biliary atresia. Universal stool-colour-card screening, as pioneered in Taiwan and Japan, demonstrates that early detection is feasible at scale and improves the age at Kasai, and it represents a model for population-level intervention. [7]
[7]Evidence, Guidelines & Regional Differences
The joint NASPGHAN and ESPGHAN guideline, published in 2017, provides the current international standard for the evaluation of cholestatic jaundice in infants, defining the conjugated bilirubin thresholds, the recommended diagnostic panel, and the algorithm for excluding biliary atresia. It supersedes and extends the earlier 2004 NASPGHAN guideline, adding the European consensus and updating the evidence base. Both guidelines stress that any infant with cholestasis needs prompt evaluation and that a normal ultrasound does not exclude biliary atresia. [1]
The START trial, published in JAMA in 2014, is the landmark randomised study of postoperative corticosteroids after Kasai portoenterostomy. It found that high-dose corticosteroids did not significantly improve the primary outcome of native-liver survival at two years compared with placebo, tempering enthusiasm for routine steroid use. The French national series of biliary atresia from 1986 to 2009 demonstrated improving outcomes over time, attributable to earlier surgery and centralised care. [6]
[5]Active controversies include whether stool-colour-card screening should be universal or targeted, the optimal steroid regimen after Kasai given the negative START trial, the role of ursodeoxycholic acid in improving bile drainage, and the timing of transplantation relative to failed Kasai. Regional differences persist in screening strategy, with universal stool-card screening established in east Asia but selective or ad-hoc detection in many Western countries where the incidence is lower. [8]
Exam Pearls
PALE for the cholestatic red flags
References
- [1]Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, McLin VA, Molleston JP, Neimark E, Ng VL, Karpen SJ Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr, 2017.PMID 27429428
- [2]Moyer V, Freese DK, Whitington PF, Olson AD, Brewer F, Colletti RB, Heyman MB Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr, 2004.PMID 15269615
- [3]Lakshminarayanan B, Davenport M Biliary atresia: A comprehensive review. J Autoimmun, 2016.PMID 27346637
- [4]Serinet MO, Wildhaber BE, Broue P, Lachaux A, Sarles J, Jacquemin E, Gauthier F, Chardot C Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics, 2009.PMID 19403492
- [5]Chardot C, Buet C, Serinet MO, Golmard JL, Lachaux A, Roquelaure B, Gottrand F, Broue P, Dabadie A, Gauthier F, Jacquemin E Improving outcomes of biliary atresia: French national series 1986-2009. J Hepatol, 2013.PMID 23402746
- [6]Bezerra JA, Spino C, Magee JC, Shneider BL, Rosenthal P, Wang KS, Erlichman J, Haber B, Hertel PM, Karpen SJ, Kerkar N, Loomes KM, Molleston JP, Murray KF, Romero R, Schwarz KB, Shepherd R, Suchy FJ, Turmelle YP, Whitington PF, Moore J, Sherker AH, Robuck PR, Sokol RJ Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: the START randomized clinical trial. JAMA, 2014.PMID 24794368
- [7]Chang MH Screening for biliary atresia. Chang Gung Med J, 2006.PMID 16924883
- [8]Matsui A Screening for biliary atresia. Pediatr Surg Int, 2017.PMID 28983697
- [9]Hukkinen M, Ruuska S, Pihlajoki M, Kyronlahti A, Pakarinen MP Long-term outcomes of biliary atresia patients surviving with their native livers. Best Pract Res Clin Gastroenterol, 2022.PMID 35331404