Paeds · fetal-neonatal-and-perinatal
Haemolytic disease of the fetus and newborn
Also known as Haemolytic disease of the newborn · Erythroblastosis fetalis · Rhesus (RhD) alloimmunisation · Alloimmune haemolytic disease · Kell alloimmunisation
Fellowship guide to haemolytic disease of the fetus and newborn: maternal IgG alloantibodies (anti-D, anti-Kell, ABO) crossing the placenta, the severity ladder from mild jaundice to hydrops, middle cerebral artery Doppler surveillance, intrauterine transfusion, and the postnatal bundle of intensive phototherapy, intravenous immunoglobulin and exchange transfusion — all anchored to antenatal anti-D prevention.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A newborn is deeply jaundiced at twelve hours of age, pale, and the mother is Rhesus-negative and never received anti-D. That is the opening scene of haemolytic disease of the fetus and newborn (HDFN), the condition in which maternal IgG antibodies directed against antigens on fetal red cells cross the placenta, bind those cells, and mark them for destruction. The result is fetal anaemia on one side of birth and neonatal hyperbilirubinaemia on the other, and the same disease can present as hydrops in utero, devastating early jaundice on the first day, or an insidious late anaemia weeks later. [3] [4]
The old name, erythroblastosis fetalis, captures the marrow's frantic response — the fetus pours out immature nucleated red cell precursors (erythroblasts) to replace the cells being haemolysed. The modern term keeps the mechanism central while signalling that the disease spans the fetal and neonatal periods, and that its severity is graded, largely preventable, and highly treatable when anticipated. [4]
Three facts shape every decision in this topic. First, prevention through anti-D prophylaxis is the single most powerful intervention, and it works upstream of all the fetal and neonatal disease. Second, the antibody identifies the mechanism and predicts the severity — RhD and anti-c cause brisk haemolysis, anti-Kell directly suppresses erythropoiesis, and ABO is usually mild. Third, the bilirubin is the threat on the neonatal side, and it is entirely preventable if anticipated and treated early. Hold those three together and the strategic frame falls into place. [1] [5]
Classification
HDFN is classified along two axes that you read together at the bedside and in the clinic. The first is the offending antibody, which sets the mechanism and the expected severity. The second is the severity ladder, judged from the degree of fetal anaemia, the presence of hydrops, and the postnatal bilirubin trajectory. [4]
RhD disease is the classic severe form: maternal anti-D, usually from a previous sensitising pregnancy or transfusion, produces brisk extravascular haemolysis that can drive the fetus into hydrops if untreated. Anti-c behaves similarly and is the next most important Rh-system antibody. Anti-Kell is the deceptive one — its antibody directly suppresses erythroid progenitors in the fetal marrow, so anaemia is disproportionate to the haemolysis and bilirubin is often deceptively low, meaning the severity is worse than the jaundice suggests. ABO incompatibility is the common mild form, usually with maternal anti-A or anti-B IgG against the infant's group A or B cells, and it often presents as early-onset jaundice without significant anaemia. [4]
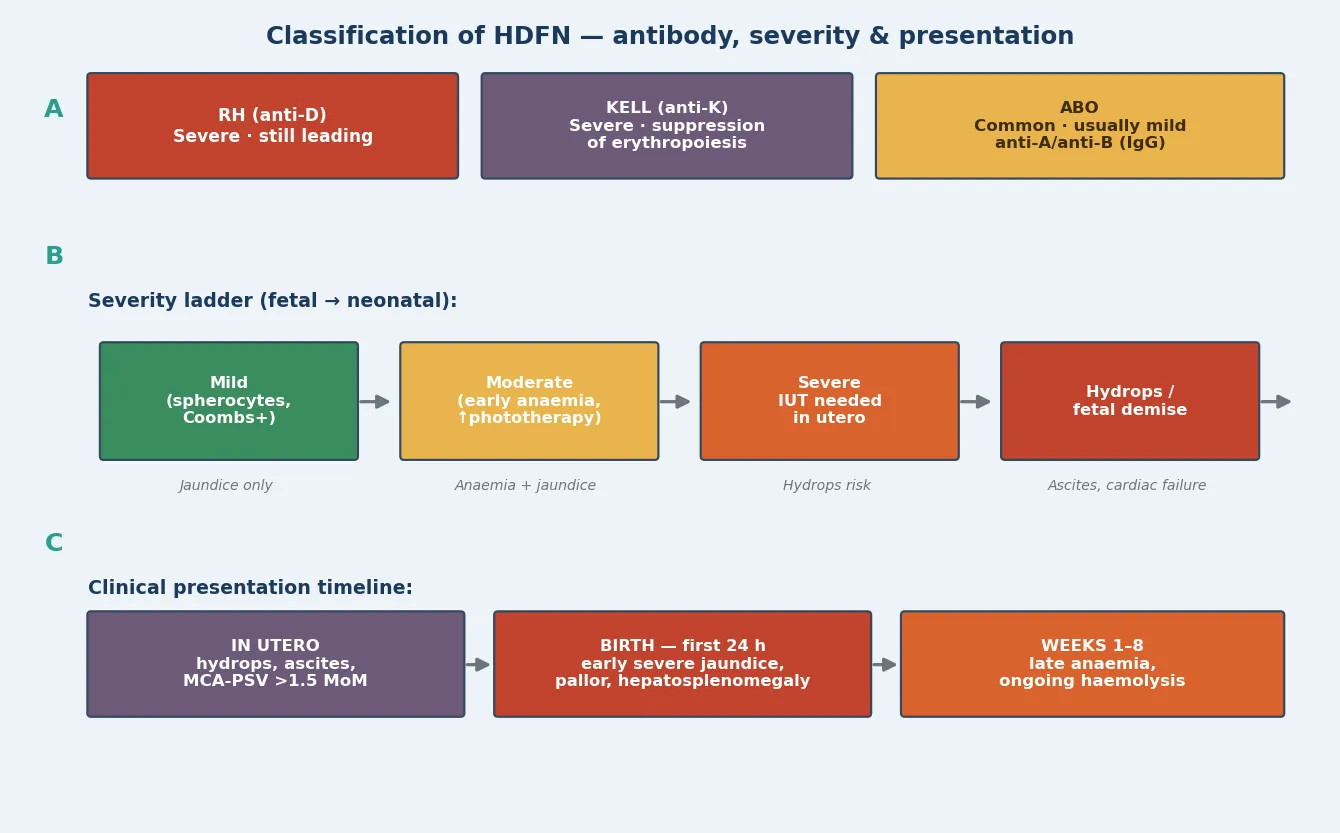
The severity ladder runs from mild disease — Coombs-positive jaundice needing phototherapy only — through moderate disease with early anaemia and a rising bilirubin, to severe disease requiring intrauterine transfusion, and finally to hydrops or fetal demise when anaemia is profound enough to cause high-output cardiac failure. Reading the antibody and the severity together is what tells you how aggressive the surveillance and treatment must be. [3] [4]
Epidemiology & Risk Factors
Before routine anti-D prophylaxis, RhD alloimmunisation affected roughly 1 per cent of pregnancies and was a leading cause of perinatal mortality. Universal antenatal and postnatal anti-D to Rh-negative mothers has reduced sensitisation dramatically, so RhD HDFN now occurs in roughly 1 in 1000 births in countries with established programmes, and most cases reflect missed prophylaxis, a sensitising event before prophylaxis, or antibodies outside the RhD system. [1] [2]
What raises and what lowers the risk of HDFN
- Rh-negative mother carrying Rh-positive fetus
- Previous sensitising pregnancy or transfusion
- Missed or late antenatal anti-D
- Sensitising event: APH, trauma, miscarriage, ectopic
- Maternal anti-c, anti-Kell or other atypical antibody
- Routine antenatal anti-D at 28 weeks
- Postnatal anti-D within 72 hours
- ABO incompatibility between mother and fetus (reduces Rh sensitisation)
- Antenatal anti-D after sensitising events
- Universal antibody screening in pregnancy
The reason ABO incompatibility between mother and fetus is itself protective against Rh sensitisation is worth knowing because it is examinable. When maternal and fetal ABO groups differ, fetal cells entering the maternal circulation are rapidly destroyed by maternal anti-A and anti-B before they can sensitise the Rh system — so ABO incompatibility acts as a natural brake on RhD alloimmunisation. [2]
Anti-Kell disease is uncommon but disproportionately severe, classically following previous transfusion, and its marrow-suppression mechanism means you must not be reassured by a bilirubin that seems modest. Setting matters too: in regions without universal anti-D programmes, or in migrant and refugee populations who may have had inconsistent antenatal care, RhD sensitisation and severe HDFN remain far more common, which is why a maternal antibody screen in pregnancy is never optional. [1] [4]
Pathophysiology
The whole disease turns on one antibody class doing one journey. Maternal IgG is the only immunoglobulin that crosses the placenta in significant quantity, transported actively across the syncytiotrophoblast from about 16 weeks' gestation via the neonatal Fc receptor. When a mother has been sensitised to a foreign red-cell antigen, her IgG alloantibody enters the fetal circulation, binds the antigen on the fetal red cell surface, and opsonises that cell. [3] [4]
The destruction that follows is predominantly extravascular. Fetal splenic macrophages bear Fc receptors that recognise the antibody-coated cell, engulf it, and break down haemoglobin into unconjugated bilirubin. The fetus clears that bilirubin through its own liver and across the placenta, which is why a sensitised fetus is anaemic but rarely jaundiced in utero — the placenta is doing the excretion. After birth, that placental clearance vanishes, and the newborn's immature conjugating liver is left to face the haemoglobin load, which is why the jaundice appears explosively in the first day. [4] [5]
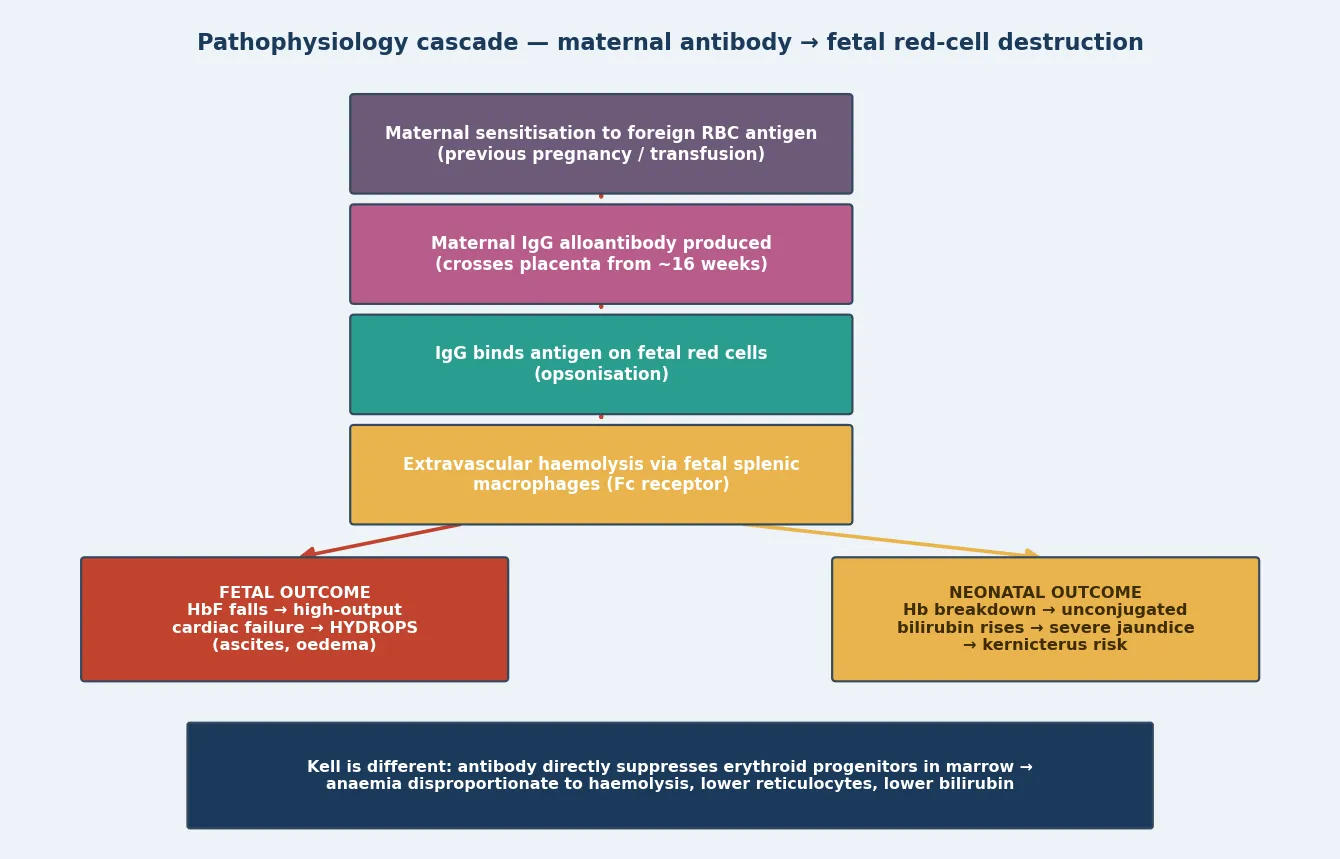
As haemolysis outstrips production, the fetal haemoglobin falls and the marrow and extramedullary sites pour out erythroblasts — hence erythroblastosis fetalis. When anaemia is severe, the heart drives a high-output state to maintain oxygen delivery, and eventually cardiac decompensation produces the transudative picture of hydrops: skin oedema, ascites, pleural and pericardial effusions. The bilirubin, liberated in the same process, is the threat on the neonatal side, because unconjugated bilirubin crosses the immature blood-brain barrier and is neurotoxic. [4] [5]
Anti-Kell follows a different path that you must distinguish. Its antibody binds the Kell antigen on erythroid progenitors in the marrow and directly suppresses erythropoiesis, so the fetus cannot mount the erythroblast response. The result is anaemia disproportionate to the visible haemolysis, a low reticulocyte count, and a bilirubin that under-represents the danger — which is why a Kell-sensitised fetus with a falling MCA-PSV trend needs IUT even when the bilirubin story seems reassuring. Knowing this mechanism changes how you read the surveillance. [4]
Clinical Presentation
HDFN shows itself differently before and after birth, and reading the presentation in each window is the skill. In utero the fetus is anaemic but not visibly jaundiced, so the presentation is ultrasound-driven: the middle cerebral artery peak systolic velocity is elevated, and as anaemia deepens the signs of hydrops appear — skin oedema, ascites, polyhydramnios from fetal swallowing failure, and pleural or pericardial effusions. By the time hydrops is visible the anaemia is profound. [3] [4]
After birth the jaundice is the signature. Unlike physiological jaundice, which appears on day two or three, haemolytic jaundice appears within the first 24 hours and climbs rapidly, and any jaundice in the first day of life demands investigation for haemolysis regardless of how well the infant looks. The infant may be pale from anaemia, with hepatosplenomegaly from extramedullary haematopoiesis, and in severe disease there is tachypnoea, poor perfusion and signs of high-output cardiac failure carried over from the fetal circulation. [5]
Presenting signs by window and severity
In utero: elevated MCA-PSV, then hydrops (oedema, ascites, effusions)
Birth to 24 h: early-onset jaundice, pallor, hepatosplenomegaly
Day 1–2: rapidly rising serum bilirubin crossing thresholds
If untreated: lethargy, hypotonia, poor suck — acute bilirubin encephalopathy
Weeks 1–8: late anaemia from ongoing low-grade haemolysis (top-up transfusion)
The time course is characteristic. Untreated, the bilirubin climbs steeply through the first days, and if it crosses the threshold for bilirubin-induced neurologic dysfunction the infant develops the signs of acute bilirubin encephalopathy: lethargy, hypotonia, poor suck, a high-pitched cry, and in the worst case opisthotonus, seizures and apnoea — the heralds of kernicterus. Beyond the neonatal period, a low-grade haemolysis may produce a late anaemia at two to eight weeks as the antibody is cleared, requiring a top-up transfusion. [5]
Some features should make you question the antibody or look for a complication. A positive direct antiglobulin test with modest bilirubin in a Kell-sensitised infant should worry you, not reassure you. A sudden fall in MCA-PSV in a hydropic fetus may signal decompensation rather than improvement. And a Coombs-negative early jaundice should send you looking for non-immune haemolysis, sepsis, or a red-cell membrane or enzyme defect rather than closing the book on HDFN. [4] [5]
Differential Diagnosis
When a newborn presents with early-onset jaundice, immune HDFN is the leading diagnosis but never the only possibility, and several mimics change management entirely. Work through the differential by the timing of onset, the direct antiglobulin test result, the rate of bilirubin rise, and the haematology. [5]
HDFN and its mimics — the tell that separates them
- Maternal antibody known, Rh-negative mother
- DAT strongly positive
- Rapid bilirubin rise, early jaundice, anaemia
- Spherocytes absent; reticulocytes high
- Mother group O, infant A or B
- DAT weakly positive or negative
- First baby can be affected; usually mild
- Spherocytes on film
- Male, Mediterranean/African/Asian heritage
- DAT negative
- Episodic haemolysis with trigger
- Bite cells, Heinz bodies
- DAT negative
- Family history of haemolysis or gallstones
- Spherocytes, ↑osmotic fragility
- Persistent jaundice
- DAT negative
- Clinical signs of infection or bruising
- Cephalhaematoma, bruising reabsorbing
- Day 2–3 onset, well infant
- Slow rise, no haemolysis
- Excluded by first-24-hour onset
Two rules govern the differential. First, a positive direct antiglobulin test confirms immune haemolysis and narrows the search to the antibody, which the maternal antibody screen and the neonatal blood group will identify. Second, a Coombs-negative early-onset jaundice with a rapid rise must still be treated as dangerous — G6PD deficiency, hereditary spherocytosis, sepsis and an enclosed haemorrhage can all produce a bilirubin trajectory that threatens the brain, and the management is driven by the bilirubin threshold, not by the label. [5] [6]
Clinical & Bedside Assessment
The bedside assessment reads the anaemia and the bilirubin risk directly. Examine the colour — early jaundice is best seen in good light by blanching the skin, progressing cephalocaudally from the face. Document pallor, perfusion, respiratory rate and effort, and feel for hepatosplenomegaly, which signals extramedullary haematopoiesis or cardiac failure. A structured cephalocaudal assessment of jaundice extent lets you gauge severity before the laboratory returns. [5]
The serum bilirubin interpreted on an hour-specific nomogram is the single most important measure on the neonatal side. Maisels' nomograms map the bilirubin against the infant's age in hours to define the risk zone and the threshold for phototherapy or exchange transfusion, adjusted for gestational age and risk factors — and haemolysis is itself a risk factor that lowers the threshold. The rate of rise, often above 8 to 10 micromoles per litre per hour in brisk haemolysis, is as informative as the absolute value. [5] [6]
On the antenatal side, the assessment reads the maternal antibody screen and the MCA-PSV together. A rising or high-titre antibody identifies the at-risk pregnancy, and the MCA-PSV, measured at the origin of the middle cerebral artery at a zero-degree angle of insonation, is the validated non-invasive test of fetal anaemia. A value above 1.5 multiples of the median for gestational age predicts moderate-to-severe anaemia and is the trigger to consider fetal blood sampling with intrauterine transfusion. [3] [4]
Investigations
The neonatal workup turns on three tests. The direct antiglobulin test (DAT, or direct Coombs) on cord or neonatal blood detects antibody bound to the infant's red cells and confirms immune haemolysis. The serum bilirubin with fractionation establishes the unconjugated hyperbilirubinaemia and its trajectory on the hour-specific nomogram. The full blood count and film reveals the anaemia, the reticulocytosis of compensatory marrow activity, and in ABO disease the spherocytes. [5] [6]
Blood-group and antibody identification on both mother and infant complete the picture: the maternal antibody screen names the offending antibody, the infant's blood group confirms incompatibility, and in many centres cell-free fetal DNA testing now determines fetal RhD status non-invasively from maternal blood, sparing an Rh-negative mother invasive procedures. The reticulocyte count and blood film distinguish the brisk haemolysis of RhD disease from the suppressed erythropoiesis of anti-Kell. [4] [5]
On the antenatal side, the investigations are serial. Maternal antibody titres or quantitative anti-D levels are monitored every two to four weeks once an antibody is identified, and when the titre or level reaches the laboratory-specific critical threshold, serial MCA-PSV Doppler is performed every one to two weeks. The MCA-PSV is the validated non-invasive test that replaced serial amniocentesis with delta-OD450, which is no longer recommended for detecting fetal anaemia. [3] [4]
In Australia and New Zealand, routine antenatal antibody screening is performed at the booking visit and at 28 weeks, anti-D is offered to all Rh-negative women antenatally and postnatally, and at-risk pregnancies are managed with serial MCA-PSV Doppler in a tertiary maternal-fetal-medicine centre, with intrauterine transfusion when the MCA-PSV exceeds 1.5 multiples of the median. Retrieval is coordinated through the regional neonatal network.
[1][4]Management — Resuscitation
Prevention is the first and most effective treatment, and it happens upstream of all the fetal and neonatal disease. Antenatal anti-D immunoglobulin, given to Rh-negative women at 28 weeks and after any sensitising event, and postnatal anti-D within 72 hours of delivering an Rh-positive infant, together suppress the maternal immune response to fetal RhD-positive cells. The Crowther Cochrane reviews established that both antenatal and postnatal anti-D substantially reduce RhD alloimmunisation, and together they have transformed a once-common lethal disease into a rarity. [1] [2]
In the delivery suite the principles are anticipation, warmth and early assessment. If the pregnancy was known to be affected, the neonatal team is present at delivery, cord blood is taken immediately for blood group, DAT, haemoglobin and bilirubin, and the infant is assessed for pallor, respiratory distress and hepatosplenomegaly. A severely affected infant may need respiratory support for the high-output cardiac state carried over from fetal life, and a profoundly anaemic or hydropic infant may require a partial exchange transfusion or top-up in the first hours. [4] [5]
Oxygen targeting is not the dominant issue here as it is in preterm lung disease, but a severely anaemic infant may need supplemental oxygen and careful fluid management to support the high-output circulation while the haemoglobin is addressed. The dominant resuscitation decisions are haematological: when to start phototherapy, when to give immunoglobulin, and when to proceed to exchange transfusion — all driven by the bilirubin threshold for the infant's age, gestation and risk factors. [5]
Management — Definitive & Stepwise
The definitive management runs along two linked ladders — an antenatal ladder that protects the fetus and a postnatal ladder that protects the newborn's brain — and the skill is escalating only when the step below fails. [4] [5]
On the antenatal side, once a clinically significant antibody is identified, the strategy is serial antibody titres or quantification, then serial middle cerebral artery Doppler from about 18 weeks, and intrauterine transfusion when the MCA-PSV exceeds 1.5 multiples of the median or hydrops develops. Mari's landmark study established that MCA-PSV detects moderate-to-severe fetal anaemia non-invasively with high sensitivity, replacing serial amniocentesis, and the SMFM guideline codified the 1.5-MoM threshold for intervention. Intrauterine transfusion is performed by experienced fetal-medicine teams at a regional centre, using O-negative cytomegalovirus-negative cross-matched blood, and it is repeated at two-to-four-week intervals guided by the predicted haemoglobin decline. [3] [4] [9]
Intravenous immunoglobulin (IVIG) for neonatal haemolysis
Dose
0.5 to 1 g per kilogram over 2 to 4 hours, repeated at 12 to 24 hours if needed
On the postnatal side, the ladder runs from intensive phototherapy, through intravenous immunoglobulin, to exchange transfusion. Intensive phototherapy with a high irradiance narrow-band blue LED source is first-line the moment the bilirubin approaches the threshold, applied to maximal exposed surface area and continued without interruption. Hansen and colleagues charted the evolution of phototherapy over sixty years, from serendipitous observation to a standardised, dosed therapy that has saved millions from death and disability — its mechanism is the photoisomerisation of bilirubin into water-soluble configurational and structural isomers that the bile and urine can clear. [5] [7]
The postnatal management ladder for HDFN
Anticipate: cord blood group, DAT, Hb, bilirubin at delivery of any sensitised infant
Intensive phototherapy at maximal surface area as the bilirubin approaches the threshold
IVIG 0.5–1 g/kg if haemolysis confirmed and bilirubin continues to rise toward exchange
Exchange transfusion if at the exchange threshold or signs of acute bilirubin encephalopathy
Top-up transfusion for late anaemia at 2–8 weeks as the antibody is cleared
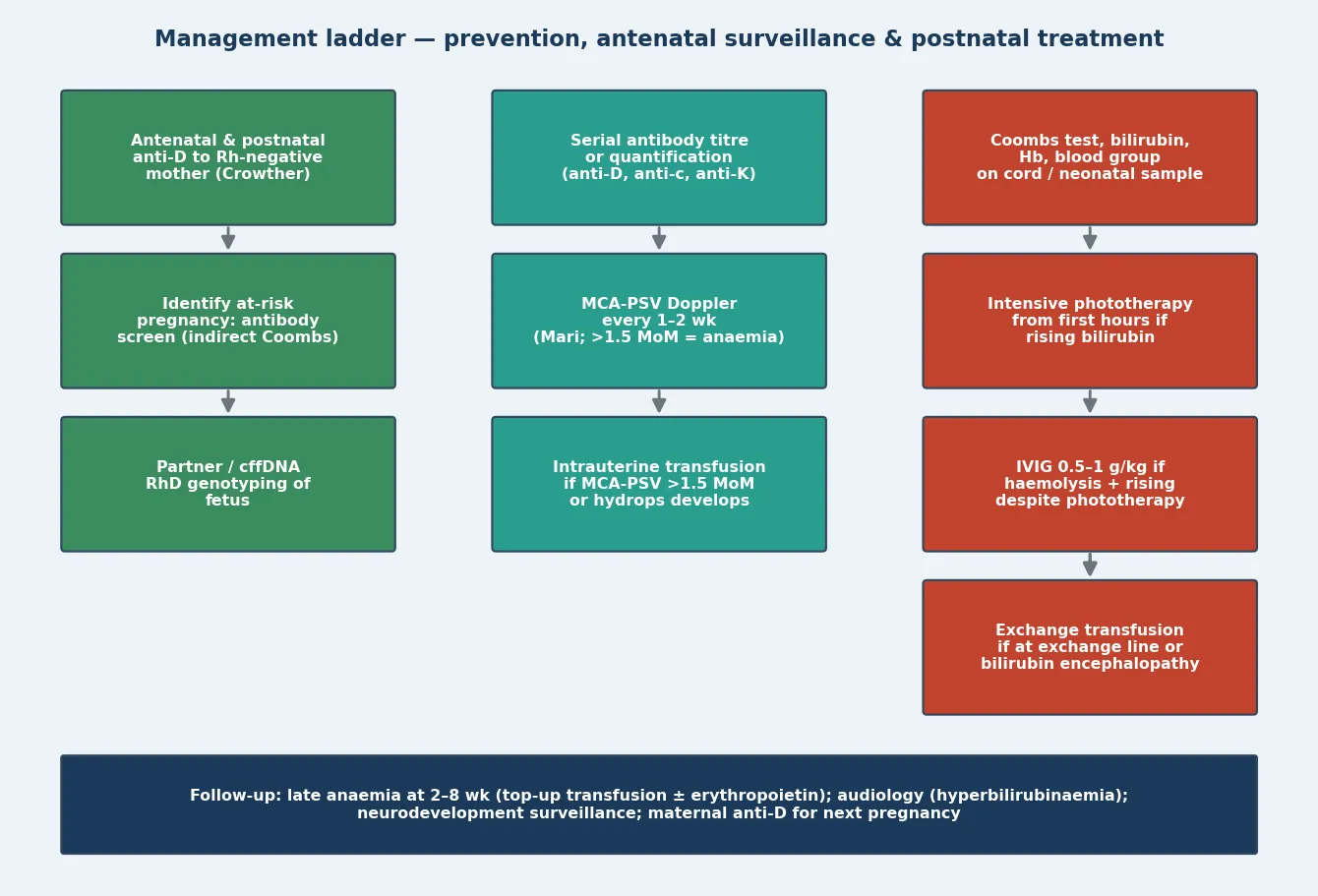
Intravenous immunoglobulin is added when haemolysis is confirmed and the bilirubin continues to rise toward the exchange threshold despite intensive phototherapy. The Zwiers Cochrane review found that IVIG reduced the need for exchange transfusion overall, though the quality of evidence is limited and the two lowest-risk-of-bias trials showed no benefit — so it is an adjunct, not a replacement for exchange transfusion when the threshold is met. Exchange transfusion is reserved for the infant whose bilirubin is at the exchange threshold or who shows signs of acute bilirubin encephalopathy, and it simultaneously removes antibody-coated red cells, bilirubin and antibody, replacing them with donor cells compatible with the maternal serum. [5] [8]
Specific Subtypes & Scenarios
The RhD-affected pregnancy is the archetype. Maternal anti-D, usually from a previous sensitising event, drives brisk haemolysis that can produce hydrops in utero. The strategy is serial antibody titres, then MCA-PSV Doppler, then intrauterine transfusion when the threshold is met, with delivery planned in a tertiary centre at 37 to 38 weeks. After birth the infant is managed on the phototherapy-IVIG-exchange ladder, and late anaemia is anticipated in the weeks that follow. [3] [4]
The anti-Kell-affected pregnancy is the one that catches out the unwary. Its antibody suppresses fetal erythropoiesis, so anaemia is disproportionate to haemolysis, reticulocytes are low, and bilirubin under-represents the severity. The surveillance is the same MCA-PSV Doppler, but the threshold to transfuse is lower because the marrow cannot compensate, and a reassuring bilirubin must never delay intervention when the MCA-PSV signals anaemia. [4]
The ABO-incompatible neonate is the common mild form. The mother is group O and the infant is group A or B, and maternal IgG anti-A or anti-B causes a usually mild haemolysis with early-onset jaundice. The DAT may be only weakly positive or even negative, spherocytes appear on the film, and anaemia is typically modest. Most infants need only phototherapy, but a minority have a rapidly rising bilirubin that requires escalation, and ABO disease can affect the first pregnancy because anti-A and anti-B IgG are naturally present. [5]
In the outborn or non-tertiary setting, the irreducible core is early recognition and early escalation. An infant with early-onset jaundice and a positive DAT needs intensive phototherapy started before transfer, a bilirubin trend documented, and urgent retrieval to a neonatal centre with exchange transfusion capability if the trajectory is steep — because the harm done by a delayed exchange in transit is preventable. [5]
Complications & Pitfalls
The acute complications of HDFN are fetal hydrops and death on one side of birth and acute bilirubin encephalopathy on the other. Hydrops reflects profound anaemia with high-output cardiac failure, and intrauterine transfusion has transformed its prognosis — Zwiers' series of 1678 procedures showed survival rising to 97 per cent in the modern era when performed in experienced centres, with procedure-related complications falling as technique improved. [4] [9]
Acute bilirubin encephalopathy and its chronic form, kernicterus, is the catastrophic complication, and it is almost entirely preventable with timely phototherapy and exchange transfusion. Unconjugated bilirubin crosses the immature blood-brain barrier and is toxic to the basal ganglia, cerebellum and brainstem nuclei. The early phase is lethargy, hypotonia and poor suck; the intermediate phase adds irritability, hypertonia, a high-pitched cry and fever; the advanced phase brings apnoea, seizures, opisthotonus and death. The chronic sequelae are the devastating and permanent kernicterus spectrum: athetoid cerebral palsy, sensorineural hearing loss, gaze palsies and intellectual disability. [5]
The common pitfalls are practical. Reassuring oneself with a modest bilirubin in a Kell-sensitised infant, delaying exchange transfusion while watching a bilirubin that is already at the threshold, failing to take cord blood at the delivery of a sensitised pregnancy, or forgetting that haemolysis lowers the phototherapy and exchange thresholds — each is avoidable and each is examinable. Document the bilirubin trajectory, the antibody, and the threshold decisions at every step. [5] [6]
Prognosis & Disposition
Outcome in HDFN is driven by the antibody, the severity at presentation, the timing of intrauterine transfusion, and the prevention of bilirubin neurotoxicity. In the era of anti-D prophylaxis, MCA-PSV surveillance and intrauterine transfusion, survival of even severely affected fetuses exceeds 95 per cent in experienced hands, and the long-term neurodevelopmental outcome for survivors is largely favourable. [9] [10]
Van Klink's cohort of children treated with intrauterine transfusions, assessed at a mean age of ten and a half years, found that health-related quality of life was broadly comparable to population norms, with some differences in parent-reported cognitive functioning and a modestly higher rate of behavioural difficulties — reassuring overall, but a reminder that severe disease carries long-term implications that deserve structured follow-up. [10]
From sensitisation to long-term follow-up
Every affected newborn needs structured follow-up: a late anaemia check at two to eight weeks, audiology because hyperbilirubinaemia threatens hearing, and neurodevelopmental surveillance for the kernicterus spectrum. The family needs counselling about anti-D for the current and future pregnancies, because prevention of sensitisation in the next pregnancy is part of the care of this one. An infant who deteriorates or cannot be managed locally is retrieved to a higher-level centre with the regional neonatal team. [1] [5]
Special Populations
The Rh-negative mother who missed anti-D is the population in whom classical RhD disease still appears. Whether through inconsistent antenatal care, a late presentation, or a sensitising event that was not covered, these pregnancies carry the full severity of unmodified disease, and the strategy is the complete bundle — antibody screening, MCA-PSV surveillance, intrauterine transfusion and the postnatal ladder. [1] [2]
In migrant and refugee populations, inconsistent antenatal care, previous transfusions in regions without antibody screening, and language and access barriers all raise the risk of undiagnosed sensitisation. A maternal antibody screen is never optional in any population, and a sensitised pregnancy needs the same surveillance and tertiary care regardless of background. [1] [4]
The anti-Kell-affected pregnancy is the deceptive special population: the antibody suppresses erythropoiesis, so anaemia is disproportionate to haemolysis and the bilirubin under-represents the danger. The MCA-PSV is the surveillance tool, and the threshold to transfuse is low, because the marrow cannot mount a compensatory response. [4]
When HDFN coexists with prematurity, the lower gestation raises the bilirubin neurotoxicity risk and lowers the exchange threshold, so an affected preterm infant needs earlier and more aggressive management. In the outborn setting, the irreducible core is early recognition, intensive phototherapy before and during transfer, and urgent retrieval to a centre with exchange capability. Family-centred care, breastfeeding support and clear communication about anti-D for the next pregnancy are part of the management, not an add-on. [5]
Evidence, Guidelines & Regional Differences
The prevention evidence comes from the Crowther Cochrane reviews of antenatal and postnatal anti-D, which together established that routine anti-D to Rh-negative mothers substantially reduces RhD alloimmunisation and the HDFN that follows. This evidence underpins the universal anti-D programmes that have made RhD disease rare in high-income settings. [1] [2]
Mari (2000, NEJM) — MCA Doppler for fetal anaemia
Population: 111 fetuses at risk of anaemia from maternal red-cell alloimmunisation, assessed before cordocentesis
Key finding
An MCA-PSV above 1.5 multiples of the median detected moderate and severe anaemia with 100% sensitivity, replacing serial amniocentesis
Practice change
MCA-PSV is the validated non-invasive test for fetal anaemia; above 1.5 MoM is the trigger for fetal blood sampling with intrauterine transfusion
The detection evidence comes from Mari's landmark NEJM study, which established that MCA-PSV above 1.5 multiples of the median detects moderate-to-severe fetal anaemia with high sensitivity, and the SMFM Clinical Guideline #8 codified this as the primary surveillance technique, retiring serial amniocentesis with delta-OD450. The intrauterine transfusion evidence comes from large single-centre series, with Zwiers' analysis of 1678 procedures showing survival of 97 per cent and falling complication rates in the modern era. [3] [4] [9]
On the neonatal side, the AAP hyperbilirubinemia guideline sets the hour-specific thresholds for phototherapy and exchange transfusion by gestational age and risk factors, with haemolysis itself a risk factor that lowers the threshold. The Maisels nomogram provides the hour-specific bilirubin percentile tracks that guide risk stratification. The Hansen phototherapy review chronicles the evolution of phototherapy into a standardised, dosed therapy, and the Zwiers IVIG Cochrane review informs the adjunctive use of immunoglobulin. [5] [6] [7] [8]
Active differences persist across regions. The timing and dosing of antenatal anti-D, the threshold for MCA-PSV-based intervention, and the role of cell-free fetal DNA RhD genotyping vary between ANZ, the United Kingdom and North America, but the convergent message is the same: prevent with anti-D, detect with antibody screen and MCA Doppler, treat the fetus with intrauterine transfusion, and treat the newborn with the phototherapy-IVIG-exchange ladder. Answer with the current guideline, name the source, and acknowledge where the evidence is still moving. [1] [4]
Exam Pearls
- Jaundice in the first 24 hours of life is haemolysis until proven otherwise — send bilirubin, DAT, full blood count and film, and blood groups. [5]
- MCA-PSV above 1.5 multiples of the median is the validated non-invasive threshold for moderate-to-severe fetal anaemia (Mari, NEJM 2000). [3]
- Antenatal and postnatal anti-D to Rh-negative mothers is the prevention that made RhD disease rare (Crowther Cochrane). [1] [2]
- Anti-Kell is deceptive: it suppresses fetal erythropoiesis, so anaemia is disproportionate to haemolysis and bilirubin under-represents severity. [4]
- ABO incompatibility is common and usually mild; mother group O, infant A or B; DAT may be weakly positive, spherocytes on film. [5]
- The direct antiglobulin test confirms immune haemolysis; a Coombs-negative rapid rise sends you to G6PD, spherocytosis or sepsis. [5]
- Plot the bilirubin on an hour-specific nomogram — haemolysis lowers the phototherapy and exchange thresholds (AAP guideline, Maisels nomogram). [5] [6]
- Intensive phototherapy is first-line; IVIG 0.5 to 1 g per kilogram is an adjunct when the bilirubin keeps rising; exchange transfusion when at threshold or encephalopathy. [7] [8]
- Intrauterine transfusion survival is 97 per cent in experienced centres (Zwiers, 1678 procedures). [9]
- Long-term outcome after IUT is largely favourable, with structured follow-up for late anaemia, audiology and neurodevelopment (van Klink). [10]
References
- [1]Crowther CA, Middleton P Anti-D administration in pregnancy for preventing Rhesus alloimmunisation. Cochrane Database Syst Rev, 2013.PMID 23450526
- [2]Crowther C, Middleton P Anti-D administration after childbirth for preventing Rhesus alloimmunisation. Cochrane Database Syst Rev, 2000.PMID 10796089
- [3]Mari G, for the Collaborative Group for Doppler Assessment of the Blood Velocity in Anemic Fetuses Noninvasive diagnosis by Doppler ultrasonography of fetal anemia due to maternal red-cell alloimmunization. N Engl J Med, 2000.PMID 10620643
- [4]Mari G, Norton ME, Stone J, Berghella V, Sciscione AC, Tate D, Schenone MH Society for Maternal-Fetal Medicine (SMFM) Clinical Guideline #8: the fetus at risk for anemia--diagnosis and management. Am J Obstet Gynecol, 2015.PMID 25824811
- [5]American Academy of Pediatrics Subcommittee on Hyperbilirubinemia Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics, 2004.PMID 15231951
- [6]Maisels MJ, Newman TB, Kaplan M A new hour-specific serum bilirubin nomogram for neonates ≥35 weeks of gestation. J Pediatr, 2021.PMID 34265340
- [7]Hansen TWR, Maisels MJ, Ebbesen F, Vreman HJ, Stevenson DK, Wong RJ, Bhutani VK Sixty years of phototherapy for neonatal jaundice - from serendipitous observation to standardized treatment and rescue for millions. J Perinatol, 2020.PMID 31420582
- [8]Zwiers C, Scheffer-Rath ME, Lopriore E, de Haas M, Liley HG Immunoglobulin for alloimmune hemolytic disease in neonates. Cochrane Database Syst Rev, 2018.PMID 29551014
- [9]Zwiers C, Lindenburg ITM, Klumper FJ, de Haas M, Oepkes D, Van Kamp IL Complications of intrauterine intravascular blood transfusion: lessons learned after 1678 procedures. Ultrasound Obstet Gynecol, 2017.PMID 27706858
- [10]van Klink JM, Lindenburg IT, Inklaar MJ, Verduin E, Koopman HM, van Kamp IL, Schonewille H, Oepkes D, Lopriore E Health-Related Quality of Life and Behavioral Functioning after Intrauterine Transfusion for Alloimmune Anemia. J Pediatr, 2015.PMID 26342721