Paeds · fetal-neonatal-and-perinatal
Neonatal acute kidney injury
Also known as Neonatal AKI · Acute renal failure in the newborn · Neonatal renal failure · Acute tubular necrosis of the newborn · neoKDIGO-defined acute kidney injury
Fellowship guide to neonatal acute kidney injury: the neonatal modified KDIGO definition and staging, the AWAKEN epidemiology, why the preterm and asphyxiated kidney is uniquely vulnerable, the prerenal–intrinsic–postrenal classification, recognition of fluid overload and electrolyte crises, the stepwise management from resuscitation to renal replacement therapy, and the lifelong CKD risk that makes post-discharge surveillance mandatory.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture a 26-week, 750-gram infant on day three of life, on mechanical ventilation for respiratory distress syndrome, now with a serum creatinine that has risen from 0.9 to 1.5 mg/dL and a urine output of 0.5 mL/kg/h. Before you dismiss this as "normal prematurity" or blame the indomethacin for the PDA, hold two ideas separate in your mind: the neonatal kidney is uniquely vulnerable, and a rising creatinine is a signal, not noise. [2]
Neonatal acute kidney injury is defined using the neonatal modified KDIGO (neoKDIGO) criteria, adapted from the 2012 KDIGO framework for the neonatal physiology where creatinine reflects maternal creatinine at birth and declines over the first weeks. [1] AKI is present when there is a serum creatinine rise of 0.3 mg/dL or more, or a rise of 50% or more from the previous lowest value, and/or when the urine output falls below 1 mL/kg/h on postnatal days 2 to 7. These criteria are deliberately structured around creatinine and urine output rather than a single threshold, because the neonatal GFR is low, tubular function is immature, and the baseline creatinine is the mother's, not the infant's. [4]
The reason this definition matters is that neonatal AKI is far more common than historically appreciated. The landmark AWAKEN study — a multicentre, multinational cohort of over 2,000 NICU admissions — found an AKI incidence of nearly 30%, with rates varying dramatically by gestational age: nearly 48% in infants below 29 weeks, 18% in those 29 to 36 weeks, and 37% in term infants. [2] More importantly, AKI was independently associated with increased mortality and longer hospital stay after adjusting for confounders — it is not a marker of illness, it is a driver of death. [2]
So the examiner's first question is always: how do you define and recognise AKI in a neonate, and why does it matter? The answer — neoKDIGO criteria, high incidence, independent mortality association — frames the entire management plan. [6]
Classification
Begin with the KDIGO stage for severity, then assign the pathophysiological category for the cause. [1]
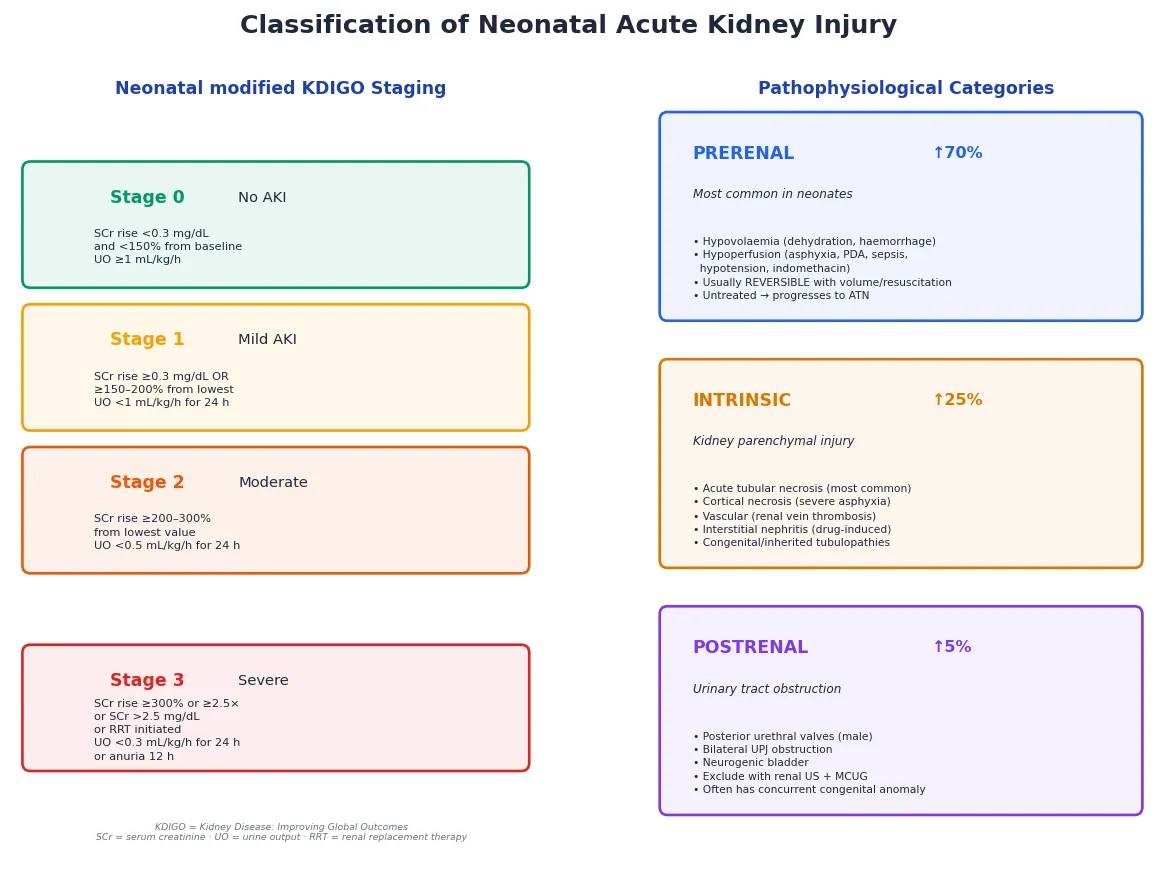
By KDIGO severity, neonatal AKI is staged from Stage 0 (no AKI) through Stage 1 (mild — creatinine rise of 0.3 mg/dL or 150–200% from baseline, or UO below 1 mL/kg/h), Stage 2 (moderate — creatinine rise 200–300% or UO below 0.5 mL/kg/h), to Stage 3 (severe — creatinine rise 300% or above, or creatinine above 2.5 mg/dL, or initiation of renal replacement therapy, or UO below 0.3 mL/kg/h for 24 hours or anuria for 12 hours). Higher stages carry proportionally worse mortality and longer hospital stay. [4]
By pathophysiological category, three mechanisms cover the ground. Prerenal AKI — the most common form in neonates — is a functional reduction in renal perfusion from hypovolaemia, hypotension, asphyxia, PDA shunting, or vasoconstrictive drugs like indomethacin and ibuprofen; the kidney is structurally intact and the injury reverses if perfusion is restored promptly. Intrinsic AKI is structural injury to the kidney parenchyma, most commonly acute tubular necrosis from prolonged ischaemia or a nephrotoxin, but also cortical necrosis after severe asphyxia, renal vein thrombosis, interstitial nephritis, and congenital or inherited tubulopathies. Postrenal AKI is obstruction of the urinary tract — posterior urethral valves in a male infant, bilateral ureteropelvic junction obstruction, neurogenic bladder, or obstructive uropathy — and must be excluded with renal ultrasound because it is often surgically correctable. [6]
Epidemiology & Risk Factors
The AWAKEN study transformed the field by providing the first robust, multicentre epidemiology of neonatal AKI. In a cohort of over 2,000 NICU admissions across four countries, the overall AKI incidence was 29.9% — nearly one in three NICU babies. [2] The incidence varies steeply by gestational age: 47.9% in infants below 29 weeks, 18.3% in the 29 to 36-week group, and 36.7% in term infants (where the higher rate reflects the sicker case mix — asphyxia, cardiac surgery, sepsis). [2]
The risk factors cluster into three groups, and the candidate must be able to recite them. Patient factors: extreme prematurity, very low or extremely low birth weight, small-for-gestational-age status, male sex, and any congenital kidney or urinary tract anomaly (which reduces nephron reserve). Perinatal factors: perinatal asphyxia with or without hypoxic-ischaemic encephalopathy, low 5-minute Apgar scores, cord blood acidosis, maternal pre-eclampsia, and chorioamnionitis. Postnatal factors: sepsis, necrotising enterocolitis, patent ductus arteriosus, respiratory distress syndrome requiring mechanical ventilation, major cardiac surgery (especially cardiopulmonary bypass), and nephrotoxic drug exposure — aminoglycosides, NSAIDs (indomethacin or ibuprofen for PDA closure), vancomycin, amphotericin, and contrast media. [6] [10]
Why these infants are vulnerable is the pathophysiology in miniature. The extreme preterm infant has the fewest nephrons because nephrogenesis is incomplete before 34 weeks; the asphyxiated infant has had a profound ischaemic insult to a kidney with low baseline GFR; the septic infant has vasoplegia and inflammatory cascades that drop perfusion further; and the cardiac surgery patient has cardiopulmonary bypass-induced inflammation and haemolysis. AKI in these groups is not a single disease but the convergence of vulnerability and insult. [6]
A temporal classification matters for surveillance: the AWAKEN study found that late-onset AKI (after day 7) occurred in about 9% of neonates and carried independently higher odds of death and longer length of stay, with unique risk factors including intubation, oligohydramnios, congenital heart disease, necrotising enterocolitis, diuretic and vasopressor exposure, and NSAID use. [3]
Pathophysiology
To understand why a neonatal kidney fails so readily, trace the nephron from its developmental origin to the cellular moment of injury. The story is one of structural vulnerability meeting an ischaemic or toxic insult. [6]
The developmental vulnerability is the foundation. Nephrogenesis — the formation of nephrons — is not complete until 34 to 36 weeks of gestation, and the full complement of nephrons (around 1 million per kidney in a term infant) is determined before birth with no capacity for regeneration. An infant born at 26 weeks has fewer than half the nephrons of a term infant, and this nephron deficit is permanent and lifelong. Superimposed on this structural immaturity is a low neonatal GFR (20–40 mL/min/1.73 m²) and immature tubular function — limited sodium reabsorption, poor concentrating ability, and impaired acid-base handling. The neonatal kidney therefore has a narrow window of reserve: it handles baseline fluid and electrolyte homeostasis, but any additional stress overwhelms it. [6]
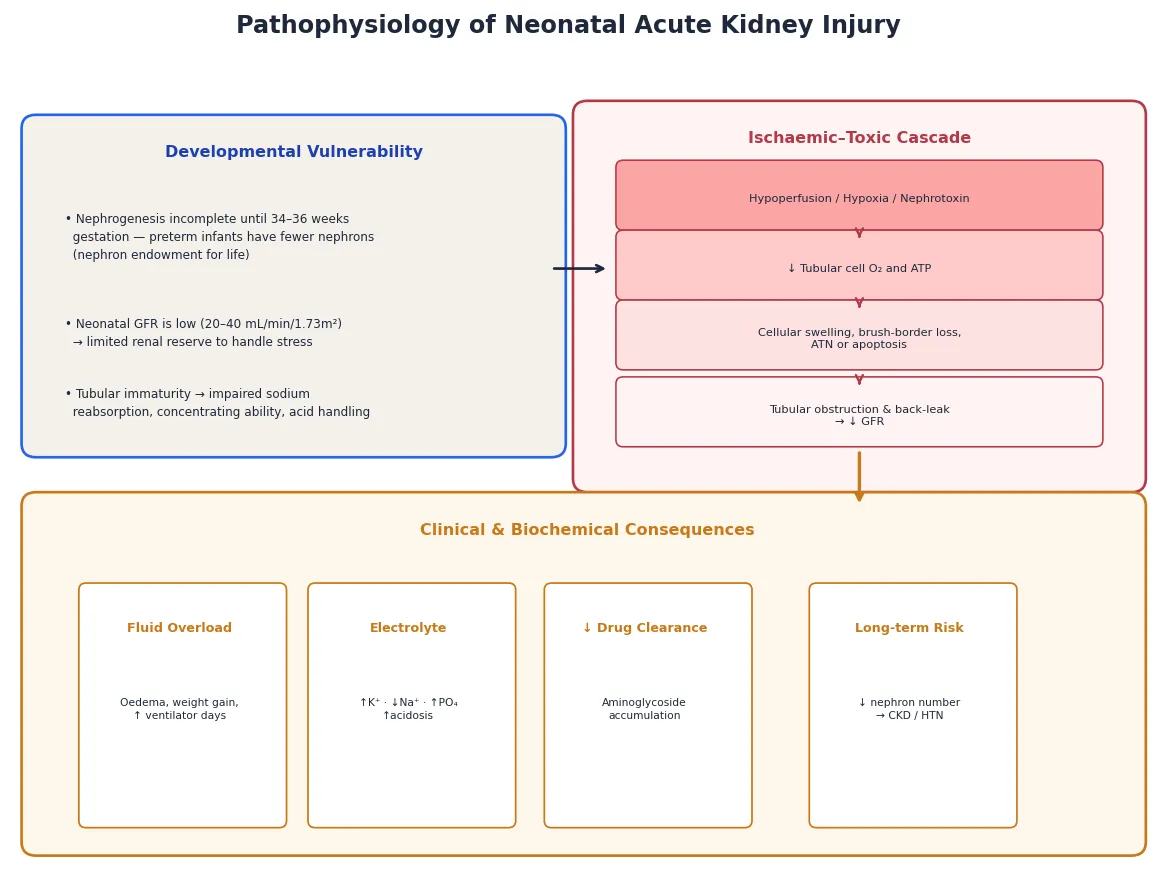
The ischaemic–toxic cascade is the mechanism of established injury. A hypoperfusion insult — from asphyxia, hypovolaemia, sepsis, or a vasoconstrictive drug — starves the tubular cells of oxygen and ATP. The cells swell, the brush border is lost, and cellular debris obstructs the tubular lumen. Tubular back-leak of filtrate across the damaged epithelium further depresses the effective GFR. If the insult is brief and the perfusion restored, the cells recover — this is prerenal AKI that has not yet become established. If the insult is prolonged, the cells die by apoptosis or necrosis, and the injury is acute tubular necrosis — an intrinsic AKI that will take days to weeks to recover even after the cause is removed. Severe and sustained asphyxia can progress to cortical necrosis, a permanent injury. [6]
One mechanism deserves emphasis because it explains the lifelong risk: every episode of AKI destroys nephrons that cannot regenerate, and the surviving nephrons undergo hyperfiltration and glomerular hypertension that accelerate chronic kidney disease over years to decades. This is why a neonate who survives an AKI episode is not simply "recovered" — they carry a permanent risk of CKD, hypertension and progressive renal decline, and they need lifelong surveillance. [6]
Clinical Presentation
The neonate with AKI shows the failing kidney through its three core functions — filtration, fluid regulation, and electrolyte balance — and the signs are the consequences of that failure. The problem is that many signs are non-specific and overlap with the underlying illness, which is why daily creatinine, weight and urine output monitoring are mandatory in the at-risk NICU infant. [2]
The urine output is the most immediately actionable sign. Oliguria (below 1 mL/kg/h) or anuria (below 0.3 mL/kg/h) may indicate prerenal, intrinsic or postrenal disease, and a bladder catheter is both diagnostic and therapeutic — it excludes obstruction and enables accurate measurement. Note that some infants with AKI (particularly aminoglycoside nephrotoxicity) are non-oliguric, so a normal urine output does not exclude AKI. [9]
The fluid and electrolyte consequences are where AKI becomes dangerous. Fluid overload presents as weight gain, oedema, and worsening respiratory status from pulmonary congestion — and in the ventilated preterm, fluid overload correlates with longer ventilation and higher mortality. Hyperkalaemia is the most dangerous electrolyte disturbance: a potassium above 6.5 mmol/L, or any ECG change (peaked T waves, widened QRS), is an emergency. Hyponatraemia is usually dilutional from water retention. Metabolic acidosis reflects the kidney's inability to excrete acid and generate bicarbonate. Hyperphosphataemia and hypocalcaemia round out the pattern. [6]
Two patterns deserve attention. The asphyxiated infant with AKI may also have HIE, cardiac dysfunction and multi-organ failure — the AKI is part of a systemic insult, and the nephrology is inseparable from the neurology and cardiology. The post-cardiac surgery infant typically develops AKI at 24 to 72 hours from the inflammatory response to cardiopulmonary bypass and the haemodynamic shifts of the post-operative period. [6]
Differential Diagnosis
The differential turns on two questions: is this prerenal, intrinsic or postrenal, and is this AKI or a congenital renal anomaly masquerading as AKI? [6]
The prerenal versus intrinsic distinction is made clinically and with urine indices. Prerenal AKI responds to a fluid challenge — the urine output recovers and the creatinine falls after 10 to 20 mL/kg of isotonic crystalloid. A fractional excretion of sodium (FENa) below 2.5% (the neonatal threshold is higher than the adult 1%) suggests prerenal conservation of sodium, while a FENa above 2.5% with a high urine sodium suggests intrinsic tubular damage and sodium wasting. A urine osmolality above 400 mOsm/kg suggests prerenal concentration, while isosthenuria (300 mOsm/kg) suggests intrinsic disease. These indices are less reliable in the preterm infant and in the diuretic- or indomethacin-exposed neonate. [9]
The postrenal causes must be actively excluded with a renal ultrasound — posterior urethral valves (a male infant with a palpable bladder and bilateral hydronephrosis), ureteropelvic junction obstruction, neurogenic bladder, or an obstructing stone. Postrenal AKI may have concurrent congenital renal dysplasia, which means the renal function may not fully recover even after relief of obstruction. [6]
The intrinsic causes beyond ATN include renal vein thrombosis (an infant with a flank mass, haematuria and thrombocytopenia, often with a history of dehydration or a clotting tendency), renal artery thrombosis (after umbilical artery catheterisation), cortical necrosis (after severe asphyxia — the creatinine plateaus high and does not recover), interstitial nephritis (drug-induced), and the congenital and inherited tubulopathies (Bartter syndrome, Fanconi syndrome, renal tubular acidosis) that present with electrolyte wasting in the first weeks. [6]
[9]Clinical & Bedside Assessment
Begin with a focused history that frames the renal risk: gestational age, birthweight and centile (SGA reduces nephron number), perinatal history (asphyxia, Apgar scores, cord pH, resuscitation), maternal history (pre-eclampsia, diabetes, oligohydramnios — which itself reflects fetal renal dysfunction), drug exposures (aminoglycosides, vancomycin, indomethacin, ibuprofen, furosemide, amphotericin, contrast), and the clinical course (sepsis, NEC, PDA, cardiac surgery, ventilation). [9] An extremely preterm infant with a PDA on indomethacin who is becoming oliguric is a very different patient from a term infant with isolated dehydration.
Accurate anthropometry sets the fluid status: weigh the baby daily and plot the trend — the expected postnatal weight loss is 5–10% over the first week, and a weight gain or a failure to lose is fluid retention. Examine for oedema (generalised, periorbital, sacral), perfusion (capillary refill, colour, pulse volume), blood pressure (hypertension suggests volume overload or renal vascular disease), and the abdomen (palpable bladder or kidneys suggest obstruction or thrombosis). [6]
Bedside monitoring is the practical backbone of AKI surveillance. Maintain strict input and output charts with a urinary catheter if output is uncertain. Check daily serum creatinine, electrolytes, urea, calcium, phosphate, magnesium and acid-base. Plot the creatinine trend — a single value is meaningless without the trajectory. In the at-risk infant, consider a renal ultrasound to exclude obstruction and assess parenchymal appearance. [9]
Finally, synthesise a one-line summary. A good example: "A 3-day-old, 26-week, 750-gram infant on indomethacin for a PDA, with a creatinine rising from 0.9 to 1.5 mg/dL and oliguria — neonatal AKI (neoKDIGO Stage 2) in an extremely preterm infant, likely prerenal from PDA-related hypoperfusion and indomethacin, exclude obstruction and reassess after volume." That sentence carries the category, the severity and the first move. [1]
Investigations
Neonatal AKI is diagnosed and surveilled at the bedside — the role of investigation is to stage the severity, classify the cause, and guide the management of complications. [4]
The cornerstone of surveillance is serial serum creatinine. In the neonate, the creatinine at birth reflects maternal renal function and declines over the first weeks as the neonatal GFR matures; a rise from the previous lowest value — by 0.3 mg/dL or 50% — is the neoKDIGO diagnostic trigger. The baseline and trend matter more than any single absolute value, and a creatinine that plateaus high or fails to decline is pathological in a neonate of any gestational age. [4]
The basic panel for every AKI episode includes serum creatinine, urea, electrolytes (sodium, potassium, chloride, bicarbonate), calcium, phosphate, magnesium, albumin, and a blood gas (for pH and acid-base). The urine studies — urine sodium, creatinine, osmolality and microscopy — help classify prerenal from intrinsic when the distinction is not clinically obvious, though they are confounded by diuretics and indomethacin. The fractional excretion of sodium (FENa) is the most useful index: below 2.5% suggests prerenal conservation, above 2.5% suggests intrinsic tubular damage. [9]
A renal ultrasound is indicated in most neonates with AKI to exclude obstruction (postrenal causes are surgically correctable), assess parenchymal echogenicity and size (echogenic, small kidneys suggest dysplasia or cortical necrosis; enlarged kidneys suggest venous thrombosis or obstruction), and identify congenital anomalies. In selected cases, a DMSA or MAG3 scan assesses cortical integrity and differential renal function, and a MCUG (micturating cystourethrogram) confirms posterior urethral valves or vesicoureteric reflux. [6]
Biomarkers — particularly urinary and plasma NGAL (neutrophil gelatinase-associated lipocalin) and cystatin C — are emerging tools that detect tubular injury earlier than creatinine, but they are not yet standard of care in most neonatal units. The candidate should know they exist and that the research is active. [6]
Know what to avoid. Do not attribute a rising creatinine to "normal prematurity" without checking the trajectory against the neoKDIGO criteria. Do not rely on a single urine output value in an uncatheterised infant. And do not delay a renal ultrasound in an anuric neonate — obstruction is correctable, and every hour of obstruction worsens the injury. [1]
Risk factors for neonatal AKI
Management — Resuscitation
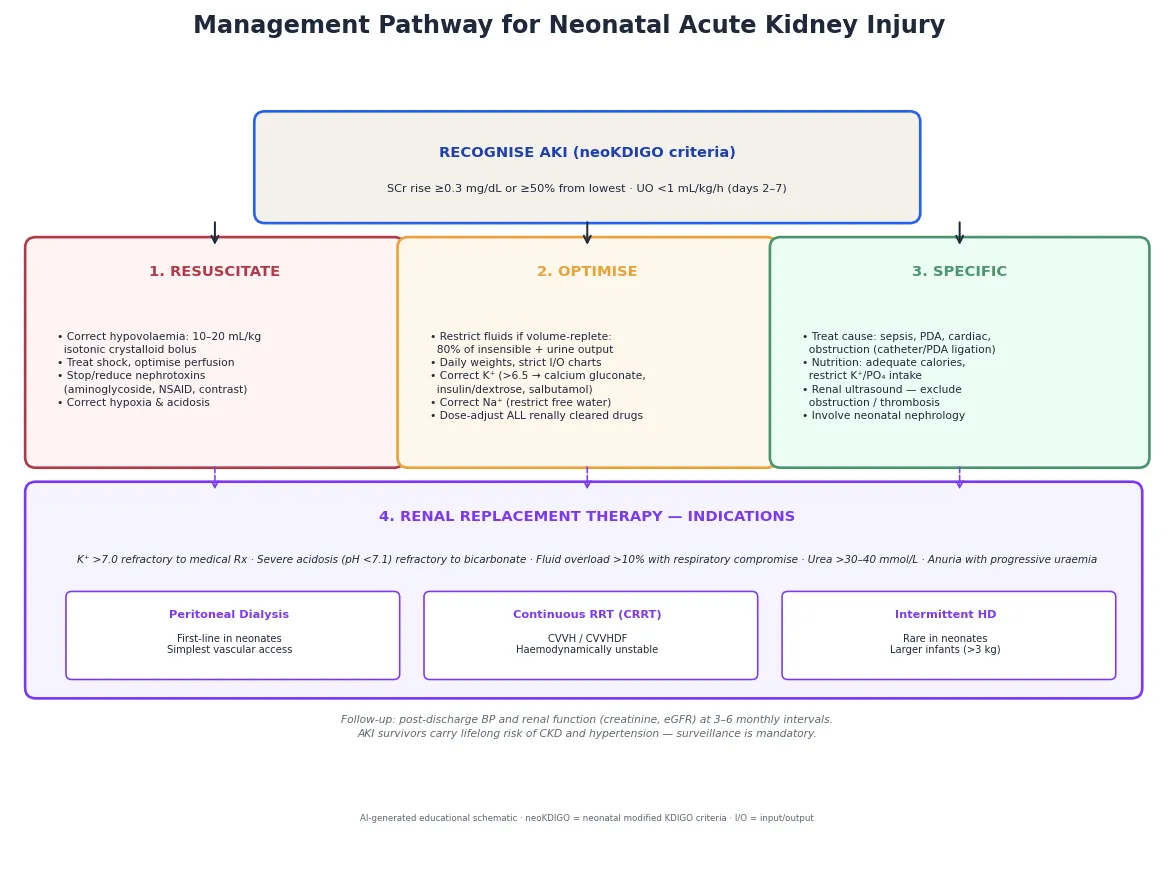
Most prerenal AKI reverses with perfusion correction — the danger is assuming that is always true, and missing the established injury that progresses. An anuric, hyperkalaemic neonate is an emergency that commands the full response. [6]
The three resuscitation priorities are restore perfusion, eliminate nephrotoxins, and treat life-threatening electrolytes. If the infant is hypovolaemic or hypotensive, give a bolus of 10–20 mL/kg of isotonic crystalloid (0.9% saline), reassess, and repeat if needed — but do not fluid-overload a infant who is already volume-replete, as that worsens pulmonary congestion and prolongs ventilation. Stop or reduce all nephrotoxic drugs: review aminoglycoside dosing (use extended-interval dosing and check levels), hold NSAIDs, use alternative antibiotics where possible, and avoid radiocontrast. [9]
Hyperkalaemia above 6.5 mmol/L, or any ECG change, is the immediate emergency. The resuscitation sequence is: calcium gluconate 10% at 0.5 mL/kg (0.11 mmol/kg) slowly intravenously to stabilise the myocardium; insulin–dextrose (0.1 units/kg insulin with about 5 mL/kg of 10% dextrose (0.5 g/kg glucose)) to shift potassium intracellularly; nebulised or intravenous salbutamol for the same purpose; and sodium bicarbonate if there is acidosis. These are temporising measures — definitive removal requires renal replacement therapy if the AKI is severe. [6]
The resuscitation errors to avoid are predictable: fluid-loading an already volume-overloaded infant (which worsens respiratory failure); continuing nephrotoxins because the team is focused on sepsis rather than the kidney; and failing to recognise that an oliguric infant on indomethacin for a PDA may have AKI that will not resolve until the PDA is addressed. [9]
Management — Definitive & Stepwise
Once stable, definitive care is a sequence you can rehearse: classify the AKI, reverse the cause, restrict fluids, correct electrolytes, dose-adjust drugs, and escalate to renal replacement therapy if refractory. [6]
- Reverse the cause. Treat hypovolaemia with fluid, sepsis with antibiotics, hypoxia with respiratory support, a haemodynamically significant PDA with closure or ligation, and obstruction with catheterisation or surgery. If a nephrotoxin is the culprit, stop it or substitute. The single most effective intervention for prerenal AKI is restoring perfusion. [9]
- Fluid management. Once volume-replete, restrict fluids to insensible losses (approximately 40 mL/kg/day for term, 60–80 mL/kg/day for preterm) plus urine output and other measured losses. Daily weights and strict I/O charts guide the prescription. A loop diuretic (furosemide 1 mg/kg) may help if the infant is oliguric and volume-overloaded, but it does not treat the AKI — it manages the fluid, and it can worsen ototoxicity and electrolyte loss. [6]
- Electrolyte management. Restrict potassium intake, use calcium resonium (or sodium polystyrene sulfonate) for enteral potassium binding, and treat hyperkalaemia as above if severe. Correct hyponatraemia by restricting free water rather than giving hypertonic saline (unless the sodium is dangerously low, below 120 mmol/L, or the infant is seizing). Restrict phosphate intake and consider phosphate binders. [6]
- Drug dose adjustment. Every renally cleared drug must be dose-adjusted to the current GFR — aminoglycosides, vancomycin, digoxin, and many antibiotics. Check levels where available. This is a patient-safety issue: a missed dose adjustment in an AKI patient causes drug accumulation and toxicity. [9]
- Renal replacement therapy. Initiate when the AKI complications are refractory to medical management: potassium above 7.0 mmol/L unresponsive to medical therapy, severe acidosis (pH below 7.1) refractory to bicarbonate, fluid overload exceeding 10% of birth weight with respiratory compromise, or progressive uraemia. The modality in neonates is peritoneal dialysis (PD) as first-line — it requires no vascular access and no anticoagulation, making it safest for the smallest infants. Continuous renal replacement therapy (CRRT) — CVVH or CVVHDF — is used in haemodynamically unstable infants with adequate vascular access, and intermittent haemodialysis is rare in neonates but possible in larger, more stable infants. [6]
The first hour of an oliguric neonate
Recognise: rising creatinine or UO <1 mL/kg/h in an at-risk infant
Check neoKDIGO criteria, daily weight trend, electrolytes, ECG
Classify: prerenal, intrinsic or postrenal
FENa, urine osmolality, renal ultrasound to exclude obstruction
Resuscitate: correct hypovolaemia with 10–20 mL/kg crystalloid
Stop nephrotoxins, optimise perfusion and oxygenation
Stabilise electrolytes: treat K⁺ >6.5 or ECG changes
Calcium gluconate → insulin/dextrose → salbutamol → bicarbonate
Escalate if refractory: fluid overload, severe hyperkalaemia, acidosis
Peritoneal dialysis first-line in neonates; CRRT if unstable
Specific Subtypes & Scenarios
AKI in the extremely preterm infant (below 29 weeks). The highest-risk group, with an AKI incidence near 48% in the AWAKEN cohort. The vulnerability is incomplete nephrogenesis and a very low GFR, compounded by respiratory distress, PDA, sepsis and nephrotoxic exposure. Management focuses on nephrotoxin minimisation, careful fluid balance, and prompt treatment of the PDA — but the preterm kidney is also more susceptible to fluid restriction (dehydration worsens perfusion), so the balance is delicate. AKI in this group is associated with higher mortality and a lifelong risk of CKD from reduced nephron number. [2]
AKI after perinatal asphyxia. The ischaemic insult injures the kidney alongside the brain and heart, and AKI is part of the multi-organ failure of HIE. Therapeutic hypothermia does not protect the kidney, and the combination of cold-induced diuresis and reduced perfusion can worsen AKI. Aminophylline or theophylline — adenosine antagonists that block the vasoconstrictive effect of adenosine on the afferent arteriole — have shown promise in randomised trials and meta-analyses for preventing AKI in asphyxiated neonates, though the evidence is still maturing in the era of therapeutic hypothermia. [7] [8]
Caffeine and AKI. The AWAKEN-derived data showed that early caffeine citrate administration in preterm infants was associated with a lower incidence of AKI — a counterintuitive but biologically plausible finding, as caffeine (a methylxanthine related to theophylline) may block adenosine-mediated renal vasoconstriction and improve renal perfusion in the preterm infant. This is a growing area of preventive nephrology. [5]
Late-onset AKI (after day 7). Defined in the AWAKEN study as AKI occurring after the first postnatal week, this affected about 9% of neonates and carried independently higher odds of death and longer length of stay. The risk factors differ from early AKI — intubation, oligohydramnios, congenital heart disease, necrotising enterocolitis, diuretic and vasopressor exposure, and NSAID use — which means surveillance must extend beyond the first week in the sick NICU infant. [3]
Cardiac surgery-associated AKI. Infants undergoing cardiopulmonary bypass for congenital heart disease have AKI rates of 30–60%, driven by the inflammatory response to bypass, haemolysis, and haemodynamic instability. The management is fluid restriction and electrolyte correction, with CRRT or PD for refractory cases. These infants also carry long-term CKD risk. [6]
Renal vein thrombosis. Presents with a flank mass, haematuria, thrombocytopenia and hypertension in an infant with risk factors (dehydration, polycythaemia, sepsis, umbilical catheter). The diagnosis is confirmed on Doppler ultrasound, and management is supportive with attention to fluid and electrolyte balance and anticoagulation in selected cases. [6]
Rural and remote setting. An oliguric, hyperkalaemic neonate born away from a NICU is an avoidable crisis — escalate and retrieve early. Establish intravenous access, give calcium for hyperkalaemia, restrict potassium-containing fluids, and transfer urgently with neonatal retrieval support. [9]
Complications & Pitfalls
Short-term, expect fluid overload (the most common and most morbid complication, correlating with longer ventilation and higher mortality), hyperkalaemia, metabolic acidosis, hyponatraemia, and the accumulation of renally cleared drugs. Long-term, the consequences are profound: every AKI episode destroys nephrons that cannot regenerate, and surviving nephrons undergo hyperfiltration and sclerosis that drive chronic kidney disease over years to decades. AKI survivors have higher rates of CKD, hypertension and progressive renal decline — which is why post-discharge surveillance is mandatory, not optional. [6]
The pitfalls are predictable, and worth naming so you avoid them. Attributing a rising creatinine to "normal prematurity" without checking the neoKDIGO-defined trajectory — the creatinine should decline, not rise, after the first days. Continuing nephrotoxic drugs because the team is focused on the sepsis or the PDA rather than the kidney. Fluid-loading an already volume-overloaded infant because the oliguria was mistaken for dehydration. Missing postrenal obstruction because a renal ultrasound was not done. Failing to dose-adjust renally cleared drugs, causing accumulation and toxicity. And discharging an AKI survivor without a renal follow-up plan, losing the window for early CKD detection. [9]
Prognosis & Disposition
Outcome is driven by the severity of the AKI (KDIGO stage), the underlying cause, the gestational age, and the promptness of treatment — not by the single highest creatinine. Most prerenal AKI reverses with perfusion correction without sequelae. The risk concentrates in the severe (Stage 3), intrinsic, asphyxia-related and cardiac surgery-associated cases, and in the extremely preterm infant with the fewest nephrons. [2]
The evidence that makes AKI important is the AWAKEN data: AKI was independently associated with a two- to four-fold increase in mortality and a longer hospital stay, even after adjusting for gestational age, severity of illness and comorbidities. [2] Late-onset AKI carries an adjusted odds ratio of 2.1 for death. [3] For long-term outcomes, the developmental nephrology evidence shows that AKI survivors carry a permanent risk of CKD and hypertension from nephron loss and hyperfiltration injury. [6]
Disposition follows the severity. A mild AKI (Stage 1) in an otherwise stable infant is managed with monitoring, nephrotoxin review and fluid balance in the NICU or special care nursery. A moderate AKI (Stage 2) requires closer monitoring, fluid restriction and electrolyte management in the NICU. A severe AKI (Stage 3) — fluid overload, refractory hyperkalaemia, severe acidosis, or needing RRT — demands full NICU support with the neonatal nephrology team involved and, in the rural setting, retrieval activated early. [9]
Post-discharge, every AKI survivor needs blood pressure and renal function (creatinine, eGFR) monitoring at 3 to 6 monthly intervals for the first years of life, because the CKD risk is lifelong and early detection of hypertension or declining function changes the trajectory. [6]
Special Populations
Extremely preterm infants (below 29 weeks) are the highest-risk group, with nearly half developing AKI and carrying the heaviest lifelong CKD burden from reduced nephron endowment. Nephrotoxin minimisation, careful fluid and electrolyte management, and a low threshold for nephrology involvement are essential, and these infants need the most rigorous post-discharge renal surveillance. [2]
Infants after perinatal asphyxia develop AKI as part of the multi-organ failure of HIE. Theophylline and aminophylline show promise for prevention, and the management is integrated with the neurology and cardiology of HIE. [7] [8]
Indigenous and socioeconomically disadvantaged families carry a higher burden of prematurity, low birthweight, gestational diabetes and reduced antenatal access — all of which raise the risk of neonatal AKI. Provide culturally safe perinatal and postnatal care, address the social determinants directly, and ensure the post-discharge renal surveillance loop does not drop. [6]
Rural and remote settings must plan for the recognition and initial management of neonatal AKI before deterioration — check daily creatinine and electrolytes in the at-risk infant, stop nephrotoxins early, and retrieve the deteriorating infant proactively. An oliguric, hyperkalaemic neonate away from a NICU is an avoidable crisis. [9]
Cardiac surgery patients are a specialised population with AKI rates of 30–60% and unique pathophysiology from cardiopulmonary bypass. These infants need coordinated neonatal, cardiac and nephrology care and long-term renal surveillance. [6]
Complex, technology-dependent infants — those with congenital kidney anomalies, chronic lung disease, or who require prolonged NICU stays — run on a specialist pathway with paediatric nephrology, urology and developmental follow-up, because the CKD and hypertension risk compounds with time. [6]
Evidence, Guidelines & Regional Differences
The foundational evidence for the epidemiology and outcomes of neonatal AKI is the AWAKEN programme: the design paper (Jetton 2016) [1] established the multicentre methodology, the main results paper (Jetton 2017) showed the 30% incidence and independent mortality association, [2] and the late-onset AKI analysis (Charlton 2019) showed that AKI after day 7 carries independently higher odds of death. [3] The AKI definition optimisation paper (Askenazi 2019) refined the neonatal creatinine thresholds by gestational age. [4]
The prevention evidence is emerging. The caffeine data (Harer 2018) showed that early caffeine citrate is associated with lower AKI rates in preterm infants. [5] The theophylline/aminophylline meta-analysis (Bhatt 2019) and the aminophylline in HIE study (Chock 2021) support adenosine antagonism for AKI prevention in asphyxiated neonates, though the evidence is still maturing in the era of therapeutic hypothermia. [7] [8]
The practice gap is documented in the survey paper (Kent 2018), which showed significant variability between neonatologists and nephrologists in AKI recognition, definition and management — the rationale for standardised neoKDIGO criteria. [9]
Two live areas: the neonatal AKI definition itself — the creatinine thresholds are being refined by gestational age, and biomarkers (NGAL, cystatin C) may complement creatinine for earlier detection; and preventive nephrology — caffeine, theophylline and nephrotoxin stewardship programmes are active research areas that may change practice in the coming years. [6]
Exam Pearls
- Neonatal AKI is defined by the neoKDIGO criteria: serum creatinine rise of 0.3 mg/dL or 50% from the previous lowest, and/or urine output below 1 mL/kg/h on days 2 to 7. [1]
- The AWAKEN study found an AKI incidence of ~30% in NICU admissions — ~48% in infants below 29 weeks — and AKI was independently associated with a 2–4× higher mortality. [2]
- The neonatal kidney is uniquely vulnerable: nephrogenesis is incomplete before 34 weeks (fewer nephrons for life), the GFR is low (20–40 mL/min/1.73 m²), and tubular function is immature. [6]
- Classify by pathophysiology: prerenal (most common, reversible, FENa below 2.5%), intrinsic (ATN, FENa above 2.5%, worst prognosis), postrenal (obstruction — exclude with ultrasound). [9]
- Hyperkalaemia >6.5 mmol/L or ECG changes → calcium gluconate, then insulin–dextrose, salbutamol, bicarbonate. [6]
- RRT indications: K⁺ above 7.0 refractory, pH below 7.1 refractory, fluid overload above 10%, progressive uraemia. Peritoneal dialysis is first-line in neonates. [6]
- Caffeine is associated with lower AKI rates in preterm infants; theophylline/aminophylline may prevent AKI in asphyxiated neonates. [5] [7]
- Late-onset AKI (after day 7) affects ~9% and carries an adjusted odds ratio of 2.1 for death — extend surveillance beyond the first week. [3]
- Dose-adjust all renally cleared drugs in AKI — patient safety issue. [9]
- AKI survivors carry lifelong CKD and hypertension risk — post-discharge BP and creatinine surveillance at 3–6 monthly intervals is mandatory. [6]
References
- [1]Jetton JG, Guillet R, Askenazi DJ, et al Assessment of Worldwide Acute Kidney Injury Epidemiology in Neonates: design of a retrospective cohort study. Frontiers in Pediatrics, 2016.PMID 27486571
- [2]Jetton JG, Boohaker LJ, Sethi SK, et al; Neonatal Kidney Collaborative Incidence and outcomes of neonatal acute kidney injury (AWAKEN): a multicentre, multinational, observational cohort study. Lancet Child and Adolescent Health, 2017.PMID 29732396
- [3]Charlton JR, Boohaker L, Askenazi D, et al; Neonatal Kidney Collaborative Late onset neonatal acute kidney injury: results from the AWAKEN Study. Pediatric Research, 2019.PMID 30546043
- [4]Askenazi D, Abitbol C, Boohaker L, et al; Neonatal Kidney Collaborative Optimizing the AKI definition during first postnatal week using Assessment of Worldwide Acute Kidney Injury Epidemiology in Neonates (AWAKEN) cohort. Pediatric Research, 2019.PMID 30643188
- [5]Harer MW, Askenazi DJ, et al Association between early caffeine citrate administration and risk of acute kidney injury in preterm infants. JAMA Pediatrics, 2018.PMID 29610830
- [6]Starr MC, Charlton JR, Guillet R Advances in neonatal acute kidney injury. Pediatrics, 2021.PMID 34599008
- [7]Bhatt GC, Gogia P, et al Theophylline and aminophylline for prevention of acute kidney injury in neonates and children: a systematic review. Archives of Disease in Childhood, 2019.PMID 30798259
- [8]Chock VY, Cho SH, et al Aminophylline for renal protection in neonatal hypoxic-ischemic encephalopathy in the era of therapeutic hypothermia. Pediatric Research, 2021.PMID 32503030
- [9]Kent AL, Charlton JR, et al Neonatal acute kidney injury: a survey of neonatologists' and nephrologists' perceptions and practice management. American Journal of Perinatology, 2018.PMID 28709164
- [10]Stoops C, Sims B, et al Neonatal acute kidney injury and the risk of intraventricular hemorrhage in the very low birth weight infant. Neonatology, 2016.PMID 27490643