Paeds · fetal-neonatal-and-perinatal
Neonatal hypotonia and neuromuscular weakness
Also known as Neonatal hypotonia and neuromuscular weakness · The floppy neonate · Neonatal hypotonia · Congenital neuromuscular weakness · The hypotonic newborn
Fellowship guide to the hypotonic neonate: the central-versus-peripheral split, the motor-unit differential, targeted genetic and electrophysiological testing, and the time-critical disease-modifying therapy for spinal muscular atrophy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the infant first: a floppy, frog-legged neonate who slips through the examiner's hands, with head lag on pull-to-sit, a scarf sign that brings the elbow across the midline, and reduced resistance to passive movement. That is hypotonia — a decrease in postural muscle tone. It is not the same as weakness, which is a loss of active force, and the distinction matters because a hypotonic infant is not always weak. The decisive bedside question is therefore not "is this baby floppy?" but "is this baby also weak?" — and that single observation reorders the whole differential. [10]
Neonatal hypotonia is the presenting feature of a long list of disorders, and the first act is to split the list into two. Central hypotonia (a problem of the brain, roughly 80% of presentations) preserves strength and reflexes because the motor unit below it is intact. Peripheral hypotonia (a problem of the motor unit — anterior horn cell, nerve, neuromuscular junction or muscle, roughly 20%) lowers tone together with strength and reflexes, because the machinery that generates force is itself failing. Spinal muscular atrophy is the commonest inherited peripheral cause, congenital myotonic dystrophy the commonest congenital muscular dystrophy seen in the neonate, and Prader-Willi syndrome a frequent central cause. [10] [4]
The split is decisive because the peripheral cohort holds the treatable, time-critical disease. Spinal muscular atrophy, untreated, kills motor neurons irreversibly and ends in fatal respiratory failure in infancy; treated before symptoms with nusinersen or gene therapy, the same infant can sit, stand and survive. So the floppy neonate is a neurology emergency, not a "wait and see" presentation. [1] [2]
Classification
The floppy neonate is classified on one axis that can be read at the bedside: central versus peripheral. Central hypotonia, the larger group, describes a brain disorder — hypoxic-ischaemic encephalopathy, chromosomal aneuploidy, Prader-Willi syndrome, an inborn error of metabolism, or benign congenital hypotonia. The motor unit beneath it works normally, so strength and reflexes are preserved and the infant is often encephalopathic, dysmorphic or seizuring. Peripheral hypotonia, the smaller but more urgent group, describes a disorder of the motor unit itself; tone, strength and reflexes fall together while the brain — and therefore alertness — is spared. [10]
The peripheral causes are then arranged by where on the motor unit the lesion sits. This anatomical ordering is the diagnostic backbone, because each level points to a discriminating test and a specific therapy. [10] [8]

Central causes are named for context. They include hypoxic-ischaemic encephalopathy, the chromosomal aneuploidies (notably Down syndrome), Prader-Willi syndrome, the inborn errors of metabolism, and a residuum of benign congenital hypotonia that is a diagnosis of exclusion. They share a normal creatine kinase, a normal electromyogram and — usually — an abnormal brain. [6] [10]
Epidemiology & Risk Factors
Most hypotonic neonates — roughly four in five — have a central cause, which is why the bedside reflex of the experienced clinician is to look first for encephalopathy, dysmorphism and seizures. The peripheral cohort is smaller but disproportionately important because it carries the genetic, time-critical disease. [10]
Spinal muscular atrophy has a birth prevalence near 1 in 10,000 live births and a carrier frequency of about 1 in 50, placing it among the commonest autosomal-recessive lethal disorders of childhood and making it the single most frequent inherited cause of neonatal hypotonia. Congenital myotonic dystrophy is the commonest congenital muscular dystrophy presenting in the neonatal period, and Prader-Willi syndrome is a recurrent central cause. [4] [6] [9]
The risk factors stratify by mechanism. For central hypotonia the determinants are perinatal asphyxia and HIE, prematurity, chromosomal anomaly, and maternal metabolic or teratogenic exposure. For congenital myotonic dystrophy the key risk factor is a maternal DMPK CTG-repeat expansion, which tends to enlarge across generations (anticipation) and produces the most severely affected infants through the maternal line — the mother may carry a mild or previously undiagnosed phenotype. Polyhydramnios and reduced fetal movements point to fetal akinesia, the common pathway of severe anterior-horn and muscle disorders and the cause of arthrogryposis. [6] [9] [10]
Pathophysiology
The central-versus-peripheral distinction is a pathophysiology distinction. In central hypotonia the motor unit is intact; tone falls because dysregulated or immature supraspinal input under-drives postural tone. Strength and reflexes are preserved because the lower motor neuron, the nerve, the junction and the muscle still generate force normally. The floppy-but-strong baby therefore has a brain problem — HIE, a chromosomal disorder, Prader-Willi syndrome — even when tone is profoundly reduced. [6] [10]
In peripheral hypotonia a component of the motor unit fails, so force generation itself is impaired and power and reflexes fall with the tone. The lesion can sit at any of four levels. Spinal muscular atrophy sits at the anterior horn cell: biallelic loss of the survival motor neuron 1 (SMN1) gene depletes SMN protein, the anterior-horn motor neurons degenerate, and denervation atrophies the muscle. The neighbouring SMN2 gene, present in variable copy number, produces a little functional SMN protein and is the chief modifier of severity — fewer copies mean earlier, more severe disease. [4] [3]
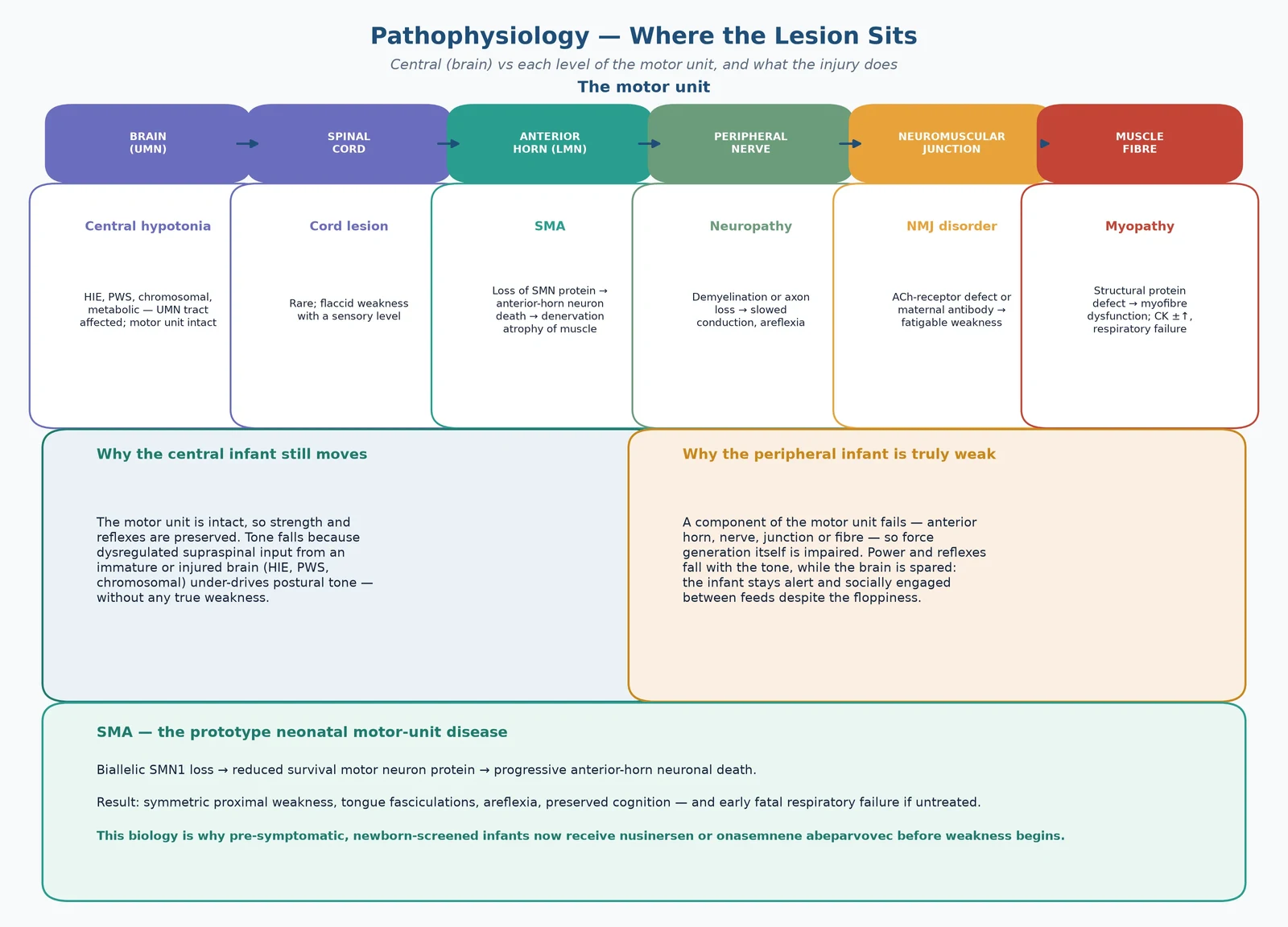
Congenital myotonic dystrophy sits at the muscle but with a systemic twist. A maternal DMPK CTG-repeat expansion generates toxic RNA that sequesters splicing regulators, producing a spliceopathy across muscle, brain, heart and respiratory drive. The result is a severely hypotonic neonate with respiratory failure, feeding difficulty and cognitive risk — and a mother who may herself be mildly affected. A neuromuscular-junction disorder (a congenital myasthenic syndrome or transient neonatal myasthenia from maternal antibody transfer) impairs synaptic transmission and produces fatigable, fluctuating weakness with ptosis. [8] [9]
The clinical payoff of this biology is therapeutic. SMA is treatable precisely because its pathophysiology is defined: nusinersen, an antisense oligonucleotide, splices SMN2 to make more functional SMN protein; onasemnogene abeparvovec delivers a functional SMN1 gene by viral vector. Both work best before the motor neurons are lost, which is why newborn screening and pre-symptomatic treatment have transformed the disease. [1] [2] [5]
Clinical Presentation
The hypotonic neonate looks floppy in a characteristic way: a frog-leg posture at rest, head lag on pull-to-sit, a scarf sign that carries the elbow across the midline, a wide popliteal angle, and reduced resistance to passive movement of the limbs. The traction response, the ventral suspension, and the assessment of axial versus appendicular tone all show the same loss of postural tone. [10]
The decisive observation is whether the infant is also weak. A central hypotonia shows normal or even brisk reflexes, preserved antigravity movement, and an infant who is often encephalopathic, dysmorphic or seizuring. A peripheral hypotonia shows genuine weakness (reduced or absent spontaneous antigravity movement), reduced or absent reflexes, and an infant who is alert and socially engaged between feeds. The floppy-but-alert baby with weak limbs and absent reflexes is the classic motor-unit presentation. [10] [8]
Red flags mark the cohort at risk of rapid deterioration. Respiratory distress or hypoventilation, a weak cry, a poor suck and swallow, tongue fasciculations, ptosis with fatigability, and fixed joint contractures at birth (arthrogryposis) each point toward a severe motor-unit disorder. Arthrogryposis is the footprint of fetal akinesia — the joints could not move in utero — and raises SMA, congenital DM1 and the congenital muscular dystrophies. [9] [10]
Differential Diagnosis
The differential is built in two moves. First, the central/peripheral split is made at the bedside on power, reflexes and alertness. Second, the level of the motor unit is identified for the peripheral group, and the specific central disorder is sought for the central group. Each move narrows the list and points to a confirming test. [10]
For the peripheral hypotonias the discriminating tests are specific. Spinal muscular atrophy is confirmed by SMN1 deletion testing with SMN2 copy number. Congenital myotonic dystrophy is confirmed by DMPK CTG-repeat testing of the infant and the mother. A congenital myasthenic syndrome and transient neonatal myasthenia are confirmed by decrement on repetitive nerve stimulation and, for the transient form, by maternal acetylcholine-receptor antibody. The congenital myopathies and muscular dystrophies are confirmed by a combination of creatine kinase, electromyography, muscle biopsy and a gene panel. [3] [8] [9]
The must-not-miss diagnoses are those whose delay harms the infant. Spinal muscular atrophy is treatable and time-critical. Congenital myotonic dystrophy carries a high anaesthetic and sedation risk and a mother who needs diagnosis. A treatable neuromuscular-junction disorder must not be missed because it responds to acetylcholinesterase inhibition or, in the transient form, resolves as the maternal antibody clears. [1] [9]
Clinical & Bedside Assessment
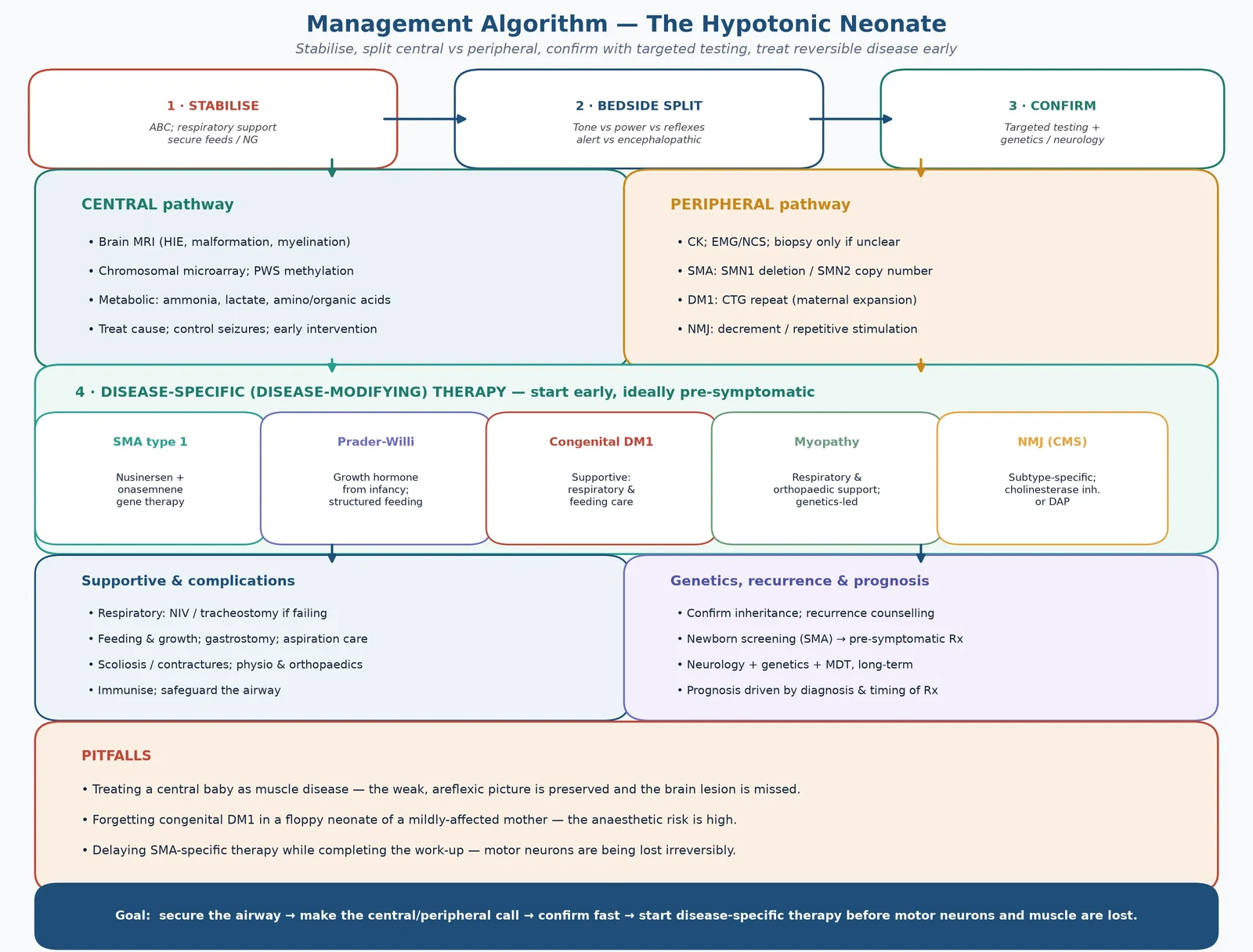
Bedside assessment has two layers — an immediate stabilisation and a focused neurological and general examination that makes the central/peripheral call. Stabilisation comes first: protect the airway, give respiratory support to the hypoventilating infant, secure safe feeding, and check the glucose, because a hypotonic infant may also be hypoglycaemic. The examination then distinguishes the cohorts. [10]
The focused examination assesses alertness; axial and appendicular tone (scarf sign, popliteal angle, head lag, ventral suspension); power by observing spontaneous and antigravity movement; the deep tendon reflexes; the tongue for fasciculations; the face and eyes for ptosis and facial weakness; the suck; the respiratory effort; and the skin and joints for dysmorphism and contractures. Three signs carry the most weight in the central/peripheral call: power, reflexes, and alertness. [10] [8]
The focused history reweights the differential. Ask about perinatal asphyxia; about polyhydramnios and reduced or absent fetal movements (a fetal-akinesia signal); about maternal weakness, myotonia, cataracts or known myasthenia gravis; about consanguinity and a family history of early infant death or neuromuscular disease; and about any antenatally diagnosed anomaly. Each answer moves the probability toward a specific cause. [6] [9] [10]
Investigations
Investigations confirm the split and the specific diagnosis. The first-line battery that confirms a peripheral cause is creatine kinase (elevated in many muscular dystrophies, often normal in SMA and the congenital myopathies), electrolytes and glucose, and brain imaging — ultrasound then MRI — for the central cohort. [8] [10]
Electromyography and nerve-conduction studies refine the level of the motor-unit lesion: denervation changes point to the anterior horn (SMA), slowed conduction to the nerve, a decrement on repetitive stimulation to the neuromuscular junction. In the neonate these studies are technically demanding and operator-dependent, and a muscle or nerve biopsy is reserved for the case where genetics and electrophysiology have not settled the diagnosis. [8]
Genetic testing now confirms most peripheral causes directly. SMN1 deletion with SMN2 copy number confirms spinal muscular atrophy and predicts severity. DMPK CTG-repeat testing of the infant and the mother confirms congenital myotonic dystrophy. A neuromuscular-junction gene panel clarifies the congenital myasthenic syndromes, and congenital-myopathy and muscular-dystrophy gene panels increasingly replace biopsy for the structural-muscle disorders. [3] [9]
The metabolic and chromosomal work-up serves the central cohort. Ammonia, lactate, amino acids and organic acids screen for inborn errors; a chromosomal microarray detects aneuploidy and copy-number variants; methylation of 15q11-q13 confirms Prader-Willi syndrome; and MRI defines the HIE pattern, malformation or disordered myelination. [6] [10]
Management — Resuscitation
Resuscitation of the hypotonic neonate follows ABCDE with two neuromuscular-specific priorities: protect the airway and support ventilation in the weak infant, and feed safely. A weak neonate hypoventilates, so the saturations are a late and misleading sign — monitor the work of breathing and the carbon dioxide, and escalate to non-invasive ventilation early. [10]
Secure the airway, give oxygen and escalate to continuous positive airway pressure or intubation if the infant is tiring, has a rising CO2, or has apnoea. Check the glucose and treat hypoglycaemia. Start safe feeding — nasogastric for the infant with a poor suck — and assess aspiration risk, because the bulbar weakness of many motor-unit disorders makes aspiration a constant threat. [9] [10]
The guiding principle is that the diagnosis drives the therapy, so establish the central/peripheral call and the specific genetic diagnosis as fast as possible. For spinal muscular atrophy this is a genuine emergency: motor neurons are lost irreversibly, and disease-modifying therapy is most effective when started before symptoms. Do not defer treatment while completing a non-essential work-up. [1] [5]
Immediate management of the hypotonic neonate
Assess and protect the airway; give respiratory support for hypoventilation — monitor CO2 and work of breathing
Check glucose; treat hypoglycaemia; start safe feeding (NG if poor suck); assess aspiration risk
Make the central/peripheral call at the bedside: power, reflexes, alertness
Send the discriminating test for the likely cohort (SMN1 for the alert weak areflexic infant; DMPK for the floppy infant of an affected mother)
Obtain CK, EMG/NCS, MRI and metabolic/microarray as the cohort demands
Refer to neurology and genetics; activate disease-modifying therapy for confirmed SMA without delay
Plan respiratory, feeding and rehabilitation support in parallel
Management — Definitive & Stepwise
Definitive management is layered: stabilise the infant, confirm the specific diagnosis, and start disease-specific therapy early. The cause-specific treatments have transformed the prognosis of spinal muscular atrophy, and they are the reason the central/peripheral call is made in minutes. [1] [3]
For spinal muscular atrophy, two disease-modifying therapies have replaced the supportive-only era. Nusinersen is an intrathecal antisense oligonucleotide that splices SMN2 to produce more functional SMN protein, and onasemnogene abeparvovec is a one-time intravenous gene therapy delivering a functional SMN1 gene by adeno-associated viral vector. The ENDEAR trial established that nusinersen improves motor function and survival in infantile-onset SMA compared with sham, and the SPR1NT trial showed that pre-symptomatic onasemnene-treated infants achieved milestones such as sitting and walking. [1] [2]
Newborn screening has shifted SMA treatment into the pre-symptomatic window, where it is most effective. The 2018 treatment algorithm directs that a screen-positive infant with two SMN2 copies (high risk of type 1) receive disease-modifying therapy immediately, while the management of higher-copy infants is individualised — because motor neurons are being lost from birth. [5]
Nusinersen (intrathecal antisense); onasemnogene abeparvovec (one-time IV AAV9 gene therapy)
Dose
Nusinersen: intrathecal loading then maintenance; onasemnene: single IV infusion — exact doses are weight/age-adjusted per local protocol
Supportive and rehabilitative care is common to all peripheral hypotonias and is not optional — it is what keeps the infant alive and functional while disease-specific therapy acts. It includes non-invasive ventilation and airway clearance for respiratory muscle weakness, gastrostomy for safe nutrition and growth, physiotherapy and orthoses for contractures, and scoliosis management. For the central cohort the therapy is cause-specific: control seizures and manage HIE, start growth hormone for Prader-Willi syndrome, and give dietary and cofactor therapy for inborn errors. [3] [6] [9]
Specific cautions attach to two diagnoses. In congenital myotonic dystrophy the anaesthetic and sedative risk is high and cardiac conduction disease needs surveillance, so sedation should be planned with anaesthesia and the heart monitored. In the neuromuscular-junction disorders, neuromuscular-blocking drugs and channel-altering agents are avoided because they can precipitate paralysis. [9]
Across Australia, New Zealand and the United Kingdom, newborn screening for spinal muscular atrophy is being progressively implemented, and nusinersen and onasemnogene abeparvovec are funded for eligible infants — with the treatment algorithm (Glascock 2018) applied so that two-SMN2-copy infants are treated before symptoms. Local eligibility criteria, funding pathways and infusion logistics vary by centre, so follow the local protocol; the biological principle — treat early, before motor-neuron loss — does not. [2] [3] [5]
Specific Subtypes & Scenarios
Spinal muscular atrophy type 1 (Werdnig-Hoffmann) is the prototype motor-unit disease and the cohort most transformed by therapy. It presents before 6 months with symmetric proximal weakness, tongue fasciculations, areflexia and preserved cognition, and it never allows independent sitting. Untreated it is fatal in infancy from respiratory failure; treated early with nusinersen or onasemnogene abeparvovec — ideally before symptoms via newborn screening — the infant can achieve motor milestones and survive. [1] [2] [3]
Congenital myotonic dystrophy presents in the neonate with severe hypotonia, respiratory failure, feeding difficulty and facial weakness, born to a mother with a DMPK CTG-repeat expansion who may be only mildly affected. There is no disease-modifying therapy; management is supportive — respiratory and feeding support, cardiac surveillance, and avoidance of anaesthetic risk — and the prognosis is guarded, with high mortality when prolonged ventilation is needed. [9]
Prader-Willi syndrome is a central hypotonia that presents with profound neonatal hypotonia, a poor suck and feeding difficulty, and hypogonadism. The diagnosis is by methylation of 15q11-q13. Early management is feeding support and growth-hormone therapy from infancy, which improves tone, body composition and development, and the cognitive and behavioural phenotype emerges later. [6] [7]
Complications & Pitfalls
The complications of neonatal neuromuscular weakness are mechanical and metabolic, and they are driven by the weakness itself. Respiratory failure from diaphragmatic and intercostal weakness is the proximate cause of death in untreated motor-unit disease. Aspiration and malnutrition follow bulbar weakness; scoliosis and contractures develop with chronic immobility; and failure to thrive accumulates from the combined energy cost of respiratory effort and poor intake. [9] [10]
The diagnostic pitfalls are the ones that delay the treatable disease. The first is labelling a peripheral hypotonia as "benign congenital hypotonia" — a diagnosis of exclusion — and discharging the weak areflexic infant without genetic testing. The second is missing congenital myotonic dystrophy in a subtly-affected mother. The third is delaying SMA-specific therapy while completing a non-essential work-up, during which motor neurons are lost irreversibly. [1] [9]
The treatment pitfalls are predictable. Giving neuromuscular-blocking or channel-altering drugs in congenital DM1 or a congenital myasthenic syndrome can precipitate paralysis. Failing to pre-empt respiratory failure — by watching saturations instead of the CO2 — costs lives. Overlooking the cardiac conduction disease of DM1 misses a preventable arrhythmia. [9]
Prognosis & Disposition
Prognosis is driven by the specific diagnosis and the timing of effective therapy — not by the degree of hypotonia at presentation. Counselling prognosis before the diagnosis is known is one of the cardinal errors, because a floppy neonate may have a self-limiting benign course, a static central disorder, or a treatable motor-unit disease with entirely different trajectories. [10]
Spinal muscular atrophy illustrates how decisively treatment changes prognosis. Untreated SMA type 1 is fatal in infancy from respiratory failure; treated before symptoms with nusinersen or onasemnogene abeparvovec, the same infant achieves motor milestones and survives. The ENDEAR trial established the nusinersen benefit, and the SPR1NT trial showed pre-symptomatic onasemnene-treated infants sitting and walking. [1] [2]
Congenital myotonic dystrophy carries a guarded prognosis. The cohort that needs prolonged ventilation beyond the neonatal period has a high mortality, and survivors carry cognitive and physical morbidity, as the Ostojić 2024 single-centre cohort quantifies. Prader-Willi syndrome has a favourable prognosis with early growth-hormone therapy and structured management, although the cognitive and behavioural phenotype persists. [6] [9]
Disposition follows severity and diagnosis. Any infant in respiratory failure or undergoing disease-modifying therapy is managed in a tertiary NICU with neurology, genetics and respiratory support. After discharge, structured neurology, genetics, respiratory and rehabilitation follow-up is essential, and the family receives recurrence-risk and prenatal counselling — which, for congenital myotonic dystrophy, includes cascade testing of the mother. [3] [5] [9]
Special Populations
The pre-symptomatic SMA infant detected by newborn screening is the population that most changes the prognosis. Because motor neurons are lost from birth, a screen-positive infant with two SMN2 copies is offered immediate disease-modifying therapy before any weakness appears — the 2018 treatment algorithm directs this — and the bar for treatment is the genetic result, not the clinical examination. [2] [5]
The late-preterm and remote or rural neonate raises retrieval considerations. A weak, hypoventilating infant needs safe transport to a tertiary centre with ventilatory capability, and the diagnostic sampling (SMN1, DMPK, CK) can be initiated at the referring hospital so that disease-modifying therapy is not delayed by geography. [10]
The infant of a mother with myotonic dystrophy or myasthenia gravis carries a specific risk. In congenital myotonic dystrophy the mother's CTG-repeat expansion is the cause, and she needs diagnosis and counselling for her own complications and for recurrence. In transient neonatal myasthenia the maternal acetylcholine-receptor antibody is transferred and the weakness resolves as the antibody clears. [9]
The neurogenetic counselling needs of the family are substantial. Spinal muscular atrophy is autosomal recessive (25% recurrence); congenital myotonic dystrophy is autosomal dominant with maternal anticipation; and Prader-Willi recurrence depends on the genetic mechanism. Cascade testing of the parents and the extended family, prenatal and preimplantation diagnosis, and access to newborn screening all form part of the family-centred plan. [4] [6] [9]
Evidence, Guidelines & Regional Differences
The evidence base for neonatal hypotonia is mature on the disease-modifying therapy of SMA and supportive for the rest. The ENDEAR trial (Finkel 2017) established that nusinersen improves motor function and survival in infantile-onset SMA compared with sham — the trial that ended the supportive-only era. The SPR1NT trial (Strauss 2022) then showed that pre-symptomatic onasemnogene abeparvovec-treated infants with three SMN2 copies achieved sitting and walking, defining the pre-symptomatic treatment benefit. [1] [2]
The consensus standard of care for SMA (Mercuri 2018) and the newborn-screening treatment algorithm (Glascock 2018) operationalise these trials into practice: a screen-positive infant is stratified by SMN2 copy number and treated before symptoms, with multidisciplinary respiratory, nutritional and orthopaedic care built around the drug. The Verhaart 2017 prevalence review anchors the epidemiology that justifies population screening. [3] [4] [5]
Newborn screening for spinal muscular atrophy is being implemented across ANZ, the UK, Europe and North America, but the pace and funding of both screening and disease-modifying therapy vary by jurisdiction. Where screening is established, pre-symptomatic treatment is the standard; where it is not, the diagnosis still rests on clinical suspicion and SMN1 testing of the weak areflexic alert infant. Equity of access to high-cost gene therapy and intrathecal nusinersen remains a live policy issue. [2] [5]
The congenital-DM1 and congenital-myopathy evidence base is supportive and registry-led, with emerging gene-specific trials. The Ostojić 2024 cohort quantifies the high early mortality of congenital DM1, and the Jungbluth 2003 congenital-myopathy review frames the classification that gene panels are now refining. Current controversies include the durability and immunogenicity of gene therapy, the question of combining nusinersen with gene therapy, and the long-term outcomes of pre-symptomatic treatment. [8] [9] [10]
Exam Pearls
Peripheral causes of the floppy neonate — 'A Nerve Joins Muscle'
References
- [1]Finkel RS; Mercuri E; Darras BT; Connolly AM; et al Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med, 2017.PMID 29091570
- [2]Strauss KA; Farrar MA; Muntoni F; Saito K; et al Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat Med, 2022.PMID 35715567
- [3]Mercuri E; Finkel RS; Muntoni F; Wirth B; et al Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord, 2018.PMID 29290580
- [4]Verhaart IEC; Robertson A; Wilson IJ; Aartsma-Rus A; et al Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis, 2017.PMID 28676062
- [5]Glascock J; Sampson J; Haidet-Phillips A; Connolly A; et al Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J Neuromuscul Dis, 2018.PMID 29614695
- [6]Cassidy SB; Schwartz S; Miller JL; Driscoll DJ Prader-Willi syndrome. Genet Med, 2012.PMID 22237428
- [7]Singh P; Mahmoud R; Gold JA; Miller JL; et al Multicentre study of maternal and neonatal outcomes in individuals with Prader-Willi syndrome. J Med Genet, 2018.PMID 29776967
- [8]Jungbluth H; Sewry CA; Muntoni F What's new in neuromuscular disorders? The congenital myopathies. Eur J Paediatr Neurol, 2003.PMID 12615171
- [9]Ostojić S; Kovačević G; Meola G; Pešović J; et al Main features and disease outcome of congenital myotonic dystrophy - experience from a single tertiary center. Neuromuscul Disord, 2024.PMID 38810326
- [10]Bodensteiner JB The evaluation of the hypotonic infant. Semin Pediatr Neurol, 2008.PMID 18342256