Paeds · fetal-neonatal-and-perinatal
Neonatal skin disorders and birthmarks
Also known as Neonatal skin lesions · Vascular birthmarks · Infantile haemangioma · Port-wine stain · Congenital melanocytic naevus · Neonatal pustular dermatoses
Fellowship guide to neonatal skin disorders and birthmarks: triaging benign and transient rashes from the syndromic and serious, the Mulliken–Glowacki biological split of vascular birthmarks, the natural history and propranolol management of infantile haemangioma, the Sturge–Weber and PHACE syndromes, giant congenital melanocytic naevi, midline lumbosacral lesions, and the counselling that reassures most families and escalates the few who need it.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A newborn's skin is doing two jobs at once: adapting to dry air after months in amniotic fluid, and carrying the marks of how it formed. Both jobs produce lesions. Most are harmless and fleeting; a small number mark a disorder that will shape the child's whole childhood. The clinician's task at the cot side is to tell those two groups apart without sending every worried parent to a specialist. [11]
Neonatal skin disorders and birthmarks span three practical groups. Benign and transient lesions — erythema toxicum neonatorum, transient neonatal pustular melanosis, milia, sebaceous hyperplasia, miliaria, dermal melanocytosis (the Mongolian spot) and harlequin colour change — are common, self-limiting, and need only explanation and reassurance. [11] [12] Birthmarks divide into vascular and pigmented. Vascular birthmarks use the biological classification of Mulliken and Glowacki, which separates proliferating vascular tumours (chiefly infantile haemangioma) from vascular malformations present at birth (port-wine stain, venous, lymphatic and arteriovenous malformations). [1] Pigmented birthmarks include the congenital melanocytic naevus and dermal melanocytosis. [13]
The single idea that organises everything is this: a lesion's distribution and natural history are more informative than its colour. A red stain on a forearm is usually nothing; the same red stain on the forehead, or a rapidly growing red plaque beside an eye, demands a work-up. A blue patch over the sacrum fades and is forgotten; a brown patch over the lower spine may tether the cord. Keep that lens and the rest follows. [4]
Classification
Start with the biological split for vascular lesions, because it drives management, then place the pigmented and transient lesions around it. [1]
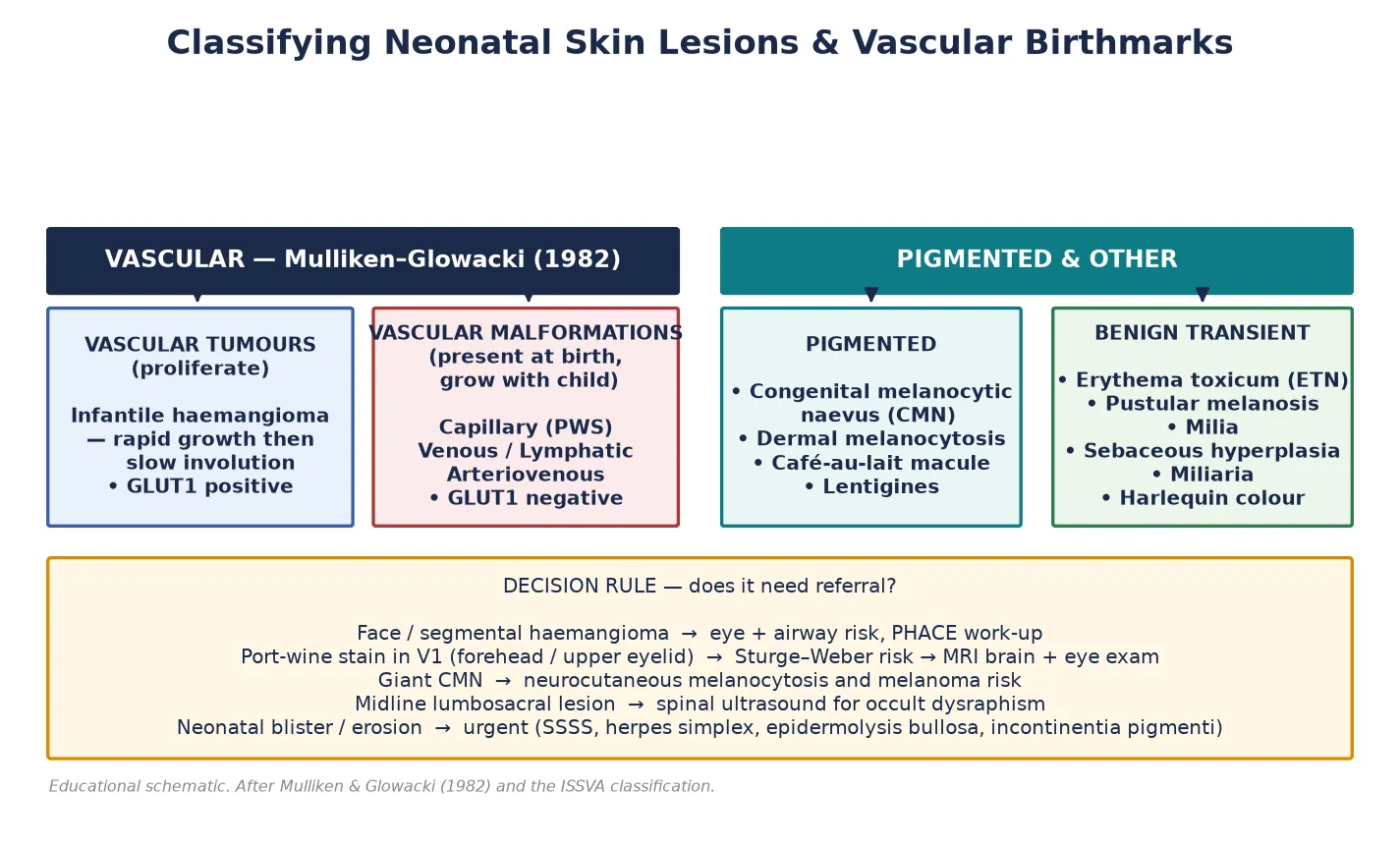
The Mulliken–Glowacki classification, published in 1982 and refined by the International Society for the Study of Vascular Anomalies (ISSVA), separates vascular anomalies by their endothelial behaviour rather than their appearance. [1] Vascular tumours are proliferative: the infantile haemangioma is the prototype, growing rapidly in the first months and then involuting over years. Vascular malformations are present at birth and grow commensurately with the child, never regressing — the port-wine stain (capillary malformation) is the neonatal prototype. The two behave differently, carry different risks, and respond to different treatments, which is why confusing them is a classic exam and clinical error. [1]
Pigmented lesions are classified by their cellular origin and size. A congenital melanocytic naevus is a benign proliferation of melanocytes present at birth; it is described as small (under 1.5 cm projected adult size), medium (1.5 to 19.9 cm), large (20 to 39.9 cm) or giant (over 40 cm), with the large and giant forms carrying the syndromic and melanoma risks. [13] Dermal melanocytosis is a common blue-grey patch of melanocytes arrested in the dermis during neural-crest migration. [10]
The transient lesions form their own group — each is a self-limiting developmental phenomenon rather than a pathology — and are best held together by their time course, as the figure below shows. [11]

Epidemiology & Risk Factors
Most newborns carry at least one benign skin lesion, and a large minority carry a birthmark — which is why the newborn skin exam is mostly a triage exercise rather than a hunt for disease. [11]
Erythema toxicum neonatorum is the commonest neonatal rash, appearing in up to half to two-thirds of term infants, typically between 24 and 48 hours of age. It is uncommon in very preterm infants in the first days, because their immune response is immature. [12] Transient neonatal pustular melanosis is less common and affects roughly 4 per cent of Black infants and under 1 per cent of white infants; unlike erythema toxicum it is present at birth. [12]
Infantile haemangioma is the commonest tumour of infancy, affecting about 4 to 5 per cent of infants by one year. The risk rises sharply in girls (a three- to fivefold female predominance), low-birth-weight and premature infants, white ethnicity, multiple gestation, and in pregnancies complicated by pre-eclampsia, placental insufficiency or advanced maternal age. [3] The recurring thread across these risks is perinatal hypoxia, which feeds the leading hypothesis that haemangioma arises from a hypoxia-driven clonal expansion of endothelial progenitor cells. [3]
Dermal melanocytosis (the Mongolian spot) is present at birth in over 80 per cent of Asian, African, Polynesian, Indigenous Australian and many Hispanic infants, and in a much smaller fraction of white infants. It usually fades in early childhood, but in some populations it persists into adulthood, which matters when an atypical spot is mistaken for a bruise. [10]
Giant congenital melanocytic naevus is rare, roughly 1 in 20,000 live births, but its syndromic burden is disproportionate. [13] The syndromic vascular lesions — PHACE and Sturge–Weber — are likewise uncommon individually, but because their cutaneous stigmata are the presenting sign, the paediatrician is usually the first to recognise them. [8] [9]
Pathophysiology
Each neonatal lesion has a mechanism, and the mechanism usually explains the natural history. Two mechanisms matter most for the exam: why an infantile haemangioma proliferates and then involutes, and why a port-wine stain on the forehead is tied to Sturge–Weber syndrome. [9]
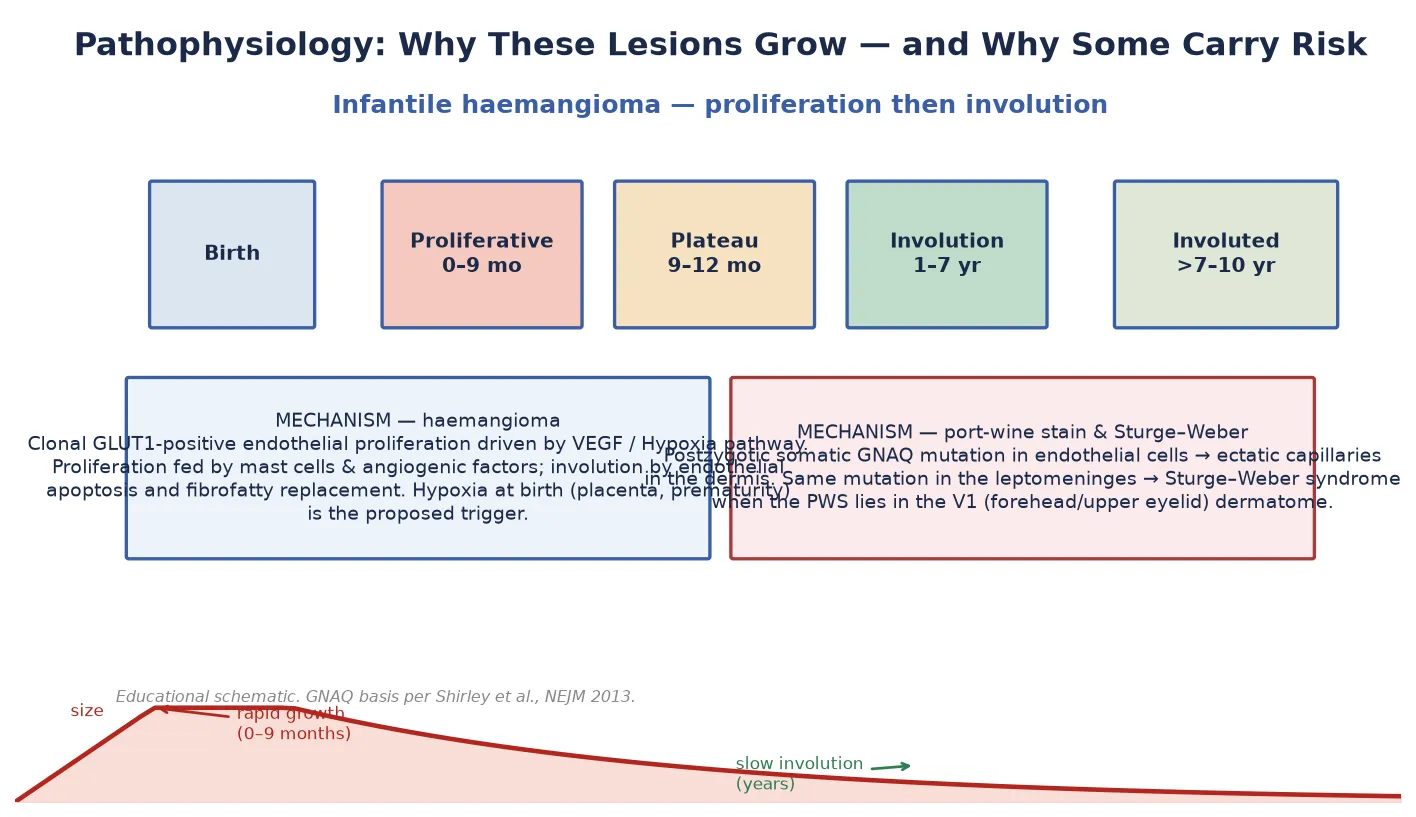
The infantile haemangioma is a benign tumour of GLUT1-positive endothelial cells — the same glucose transporter found on placental tissue, which is why the placental-embolic and hypoxia theories compete to explain its origin. During the proliferative phase (birth to roughly 9 months) the lesion expands on angiogenic signals, chiefly vascular endothelial growth factor (VEGF), supported by mast cells and endothelial progenitor cells. [3] In the involuting phase (1 to 7 years) endothelial apoptosis takes over and the vascular tissue is replaced by fibrofatty stroma, which is why a fully involuted haemangioma leaves a pale, sometimes wrinkled or telangiectatic patch. [4]
The port-wine stain has a completely different biology. It is a congenital capillary malformation — ectatic, fully formed capillaries in the superficial dermis — and the cause is now known. Shirley and colleagues showed in 2013 that a postzygotic (mosaic) activating mutation in GNAQ underlies both the isolated port-wine stain and Sturge–Weber syndrome. [9] When the mutation arises in the embryonic cell population destined to form the skin of the forehead and upper eyelid (the V1 dermatome) and the overlying leptomeninges, the child develops both the cutaneous stain and the leptomeningeal angiomatosis that defines Sturge–Weber. That is why the distribution of the stain — not its colour — predicts the neurological risk. [9]
The transient lesions each have a simple developmental mechanism. Erythema toxicum is an eosinophil-driven follicular reaction to the new world of skin flora. [12] Milia and sebaceous hyperplasia are driven by maternal and maternal-placental androgens acting on sebaceous units. Miliaria is obstruction of the sweat duct in a warm or over-wrapped baby. Harlequin colour change reflects immature autonomic vasomotor control producing a transient, sharp midline colour split. Dermal melanocytosis is melanocytes arrested in the dermis during their neural-crest migration, which is why it is blue (Tyndall effect) and usually fades as the cells disperse. [10] [11]
Giant congenital melanocytic naevi arise from an early embryonic NRAS or BRAF mutation in a melanocyte precursor; when melanocytes seed the leptomeninges the result is neurocutaneous melanocytosis, and the same deranged biology carries a small but real lifetime melanoma risk. [13]
Clinical Presentation
What you see at the bedside usually answers the triage question before any test. Recognise each common lesion by its morphology, site, and timing, then look deliberately for the distributions that change the plan. [11]
Erythema toxicum neonatorum appears in the first one to two days as erythematous macules and papules, each topped by a central 1 to 3 mm papule or pustule, scattered over the trunk, face and proximal limbs and sparring the palms and soles. The lesions migrate and evolve hour to hour, which is itself reassuring. [12]
Transient neonatal pustular melanosis is present at birth as flaccid, non-erythematous vesicopustules that rupture within a day or two to leave a collarette of scale around a small brown pigmented macule. Crucially it can affect the palms and soles, and it leaves pigmentation — two features that distinguish it from erythema toxicum. [12]
Milia are 1 mm firm white papules on the nose, cheeks and forehead — tiny epidermoid cysts. Sebaceous hyperplasia is a carpet of minute yellow papules on the nose from over-active sebaceous glands. Both are maternal-androgen driven and fade in weeks. [11]
Dermal melanocytosis is a blue-grey, slightly mottled macule most often over the sacrum, buttocks, flanks or shoulders, present at birth and non-tender. Miliaria is fine clear (crystallina) or red (rubra) papules in a hot, over-wrapped baby. Harlequin colour change is a dramatic but benign transient flush down one half of the body with sharp pallor of the other, lasting seconds to minutes in the first week. [11]
The infantile haemangioma is usually absent or a faint mark at birth, appears in the first two weeks, and proliferates fast. Superficial lesions are a bright red, lobulated ("strawberry") plaque; deep lesions are a warm bluish mass with overlying telangiectasia; mixed lesions show both. [3] A port-wine stain is a pink-red to deep purple, sharply demarcated macule present at birth in a dermatomal distribution, fixed and not fading; over years it thickens and may develop vascular blebs. [9]
The congenital melanocytic naevus is a brown, often darkly pigmented and sometimes hairy plaque present at birth; a giant naevus covers a large area such as the trunk or a limb in a garment-like or bathing-suit distribution, often with satellite lesions. [13]
Differential Diagnosis
The differentials worth rehearsing are the ones where a benign lesion imitates a dangerous one. Each has a discriminating feature you can name at the bedside. [12]
For vascular lesions, the key distinctions are between the infantile haemangioma (absent at birth, proliferates, GLUT1 positive) and the congenital haemangioma (fully grown at birth; the rapidly involuting RICH regresses in the first year, the non-involuting NICH persists), and between a haemangioma and a vascular malformation (present at birth, GLUT1 negative). [1] A pyogenic granuloma is a friable, easily bleeding red papule in an older infant. A kaposiform haemangioendothelioma and tufted angioma are the lesions behind the Kasabach–Merritt phenomenon — a rapidly enlarging vascular mass with consumptive thrombocytopenia, a true emergency. [4]
A blue-grey patch in a neonate has a short, important differential. Dermal melanocytosis fades and sits in the typical sacral or flank sites; a blue naevus is a smaller, stable, usually acral blue papule; a bruise from birth trauma is tender, evolves in colour and resolves. The trap runs both ways: a Mongolian spot can be mistaken for a bruise and trigger a safeguarding concern, and a real bruise on a non-mobile infant can be dismissed as a birthmark — so document size, site and evolution carefully. [10]
The neonatal blistering disorders are a small but dangerous group: staphylococcal scalded skin syndrome (diffuse tender erythema and peeling around flexures and orifices), herpes simplex (clustered vesicles, often scalp or eye, ill infant), epidermolysis bullosa (blisters at sites of friction, present from birth), and incontinentia pigmenti (a linear blistering rash in a female neonate that later evolves through warty and pigmented stages). [11]
Finally, a port-wine stain must be separated from the much commoner salmon patch (naevus simplex) — a fading pink stain of the eyelid ("angel kiss"), glabella or nuchal area that blanches and resolves over months — and from an early haemangioma, which will proliferate. [11]
Clinical & Bedside Assessment
A focused newborn skin exam is the single most useful investigation for these lesions. Do it in good light, with the infant fully exposed, and map what you see. [4]
Inspect every region in turn — scalp, face, neck, trunk, folds, genitals, palms, soles and the midline of the back. Blanch erythematous lesions with a glass slide or finger to separate a blanching vascular stain from a fixed port-wine stain or a petechial lesion. Palpate for warmth (suggests proliferative haemangioma), induration or tenderness (suggests infection or ulceration), and subcutaneous mass (a deep haemangioma or lipoma). Measure any lesion with a ruler and photograph it with a size reference, because serial photographs are how you confirm a haemangioma is proliferating or involuting. [4]
Then look deliberately for the four distributions that change management. Examine the face for a segmental haemangioma over 5 cm (PHACE), the forehead and upper eyelid for a port-wine stain (Sturge–Weber), the midline lower back for a dimple, lipoma, hair tuft, tag or deviated gluteal cleft (occult dysraphism), and the whole skin to count cutaneous haemangiomas (five or more triggers hepatic screening). [8] [9]
A useful bedside test when a pustule is hard to characterise is a Wright or Giemsa stain of pustule contents: erythma toxicum shows eosinophils, transient neonatal pustular melanosis shows neutrophils and melanophages, and a bacterial or herpetic pustulosis shows the organism on Gram stain or HSV PCR. [12] A Wood's lamp helps to delineate café-au-lait macules and subtle pigmentation, though it is rarely needed in the newborn period.
Construct a one-line problem representation at the cot side. A well term infant with a migratory erythematous pustular rash sparing the palms is a clinical erythema toxicum needing no tests. A six-week-old with a rapidly enlarging red plaque on the upper eyelid is a periocular haemangioma needing same-week ophthalmology. [4]
Investigations
Most neonatal skin lesions are diagnosed clinically and need no investigation at all — a point worth stating explicitly because over-investigation of the benign majority is itself a failure of care. [11]
When a lesion is uncertain, a Wright or Giemsa stain of pustule contents, a bacterial culture, and an HSV PCR distinguish the benign pustular rashes from staphylococcal or herpetic disease. These are the only "tests" most transient lesions ever require, and only when the infant is unwell or the morphology is atypical. [12]
The syndromic and serious lesions each carry a defined work-up. A large or segmental facial haemangioma over 5 cm prompts a PHACE screen: brain and cervical MRI and magnetic resonance angiography (MRA) for posterior fossa malformation and arterial cerebrovascular anomalies, an echocardiogram and aortic-arch imaging for cardiac and aortic coarctation, and an ophthalmology review for eye anomalies. [8] A port-wine stain in the V1 distribution (forehead or upper eyelid) prompts gadolinium-enhanced brain MRI to look for leptomeningeal angiomatosis and an ophthalmology review for glaucoma; the timing is early infancy, because glaucoma and seizures can declare in the first year. [9]
A midline lumbosacral lesion prompts a spinal ultrasound in the first three to six months (before the posterior vertebral elements ossify and obscure the view), with MRI reserved for an abnormal ultrasound or an older infant, looking for a tethered cord, spinal lipoma or occult dysraphism. [4]
When there are five or more cutaneous haemangiomas, perform an abdominal ultrasound to look for hepatic haemangioma, and check a thyroid function panel because large hepatic haemangiomas produce type 3 iodothyronine deiodinase and cause consumptive hypothyroidism. [4]
A GLUT1 immunostain on biopsy is occasionally used to confirm an infantile haemangioma when the distinction from a vascular malformation or congenital haemangioma is unclear — GLUT1 is positive in the infantile haemangioma and negative in malformations and congenital haemangiomas. [1] Biopsy is otherwise reserved for a lesion behaving atypically, such as a rapidly enlarging mass with thrombocytopenia (suspect kaposiform haemangioendothelioma). [4]
Management — Resuscitation
Most neonatal skin lesions need no resuscitation at all — but a small group are time-critical, and recognising them at the cot side is the job. [4]
The subglottic or airway haemangioma presents with progressive stridor, usually worse with crying or supine positioning and often in the first two months of life, frequently alongside a cutaneous "beard-distribution" haemangioma. This is an emergency: admit, start oral propranolol early, involve ENT and anaesthesia for airway assessment, and consider systemic corticosteroids. [5]
The Kasabach–Merritt phenomenon — a rapidly enlarging vascular lesion with consumptive thrombocytopenia, petechiae and a falling platelet count — is a haematological emergency managed in a tertiary paediatric unit with supportive care, platelet and coagulation support as needed, and specific therapy of the underlying kaposiform haemangioendothelioma or tufted angioma. [4]
A neonatal blistering or erosive disorder that is widespread, painful, or accompanied by systemic illness needs same-day paediatric and dermatology assessment with swabs for HSV PCR and bacterial culture. Staphylococcal scalded skin syndrome, disseminated herpes simplex and the inherited blistering disorders all present this way, and each has a different emergency treatment. [11]
An ulcerated, bleeding haemangioma needs local wound care, haemostasis, analgesia, and prompt initiation of oral propranolol to arrest proliferation and allow healing, with a low threshold for specialist review. [4]
Avoid the opposite error: treating the benign majority. Erythema toxicum, milia and sebaceous hyperplasia need no topical agents, no antibiotics and no laboratory tests — giving them causes harm, cost, and parental anxiety. [11]
Management — Definitive & Stepwise
Definitive management splits cleanly by diagnosis. The transient lesions are managed by reassurance alone; the vascular, pigmented and syndromic lesions follow defined ladders. [4]
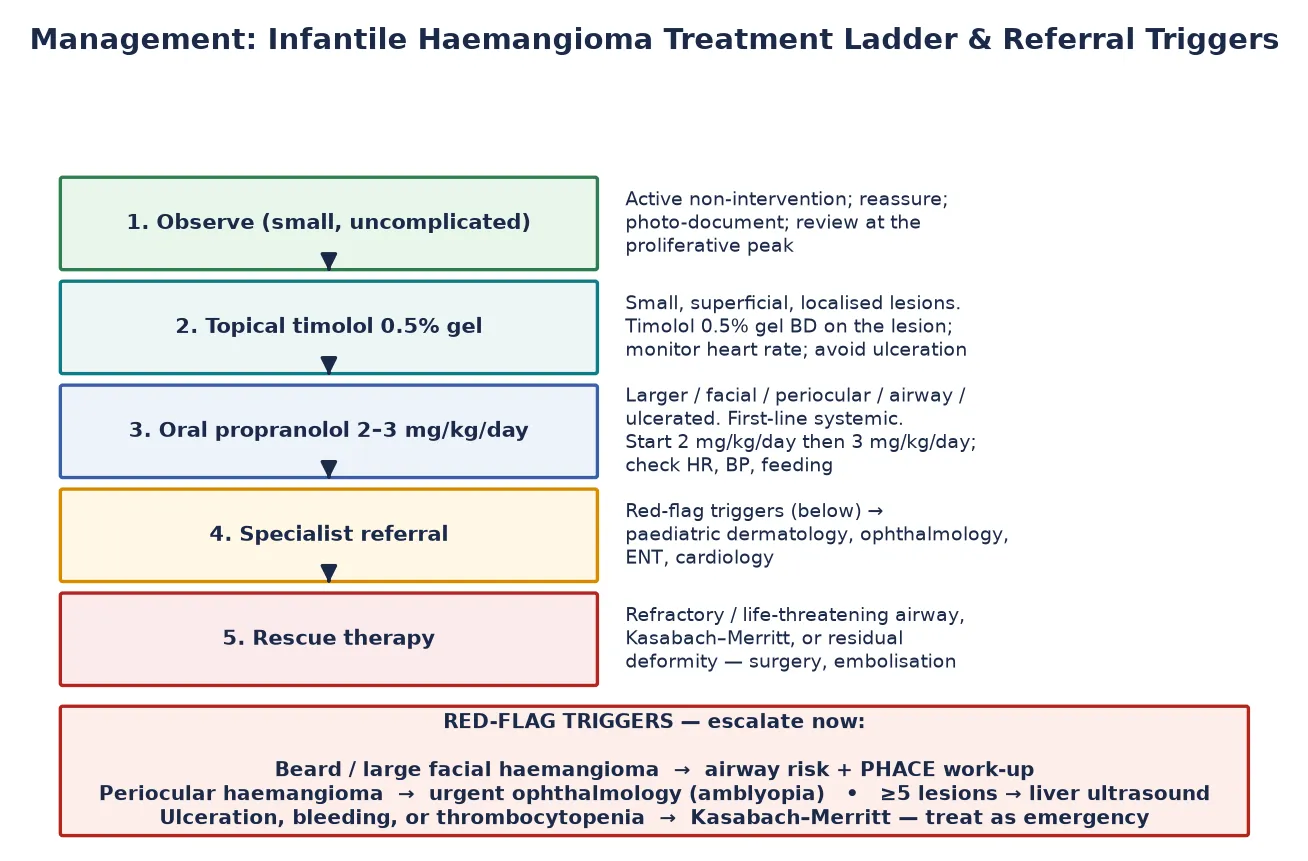
For the infantile haemangioma, the AAP clinical report frames a stepwise ladder. [4] A small, uncomplicated lesion on the trunk is observed with serial photographs through the proliferative peak. A small, superficial lesion in a cosmetically sensitive but low-risk site is treated with topical timolol 0.5% gel applied to the lesion once or twice daily, with heart-rate monitoring. [4] A lesion that is large, segmental, facial, periocular, airway-related, ulcerated, or impairing function is treated with oral propranolol 2 to 3 mg/kg/day in divided doses, continued through proliferation to roughly 12 to 18 months of age. [4] [5]
Oral propranolol for infantile haemangioma
Dose
Start 2 mg/kg/day in 2–3 divided doses, titrate to 3 mg/kg/day
Propranolol was introduced to this field by serendipity in 2008, when Léauté-Labrèze and colleagues observed haemangioma regression in infants treated with propranolol for cardiac indications. [2] It is now first-line systemic therapy, supported by the consensus conference of Drolet and colleagues, who set out initiation, dose, monitoring and contraindications. [5] Baselga and colleagues confirmed efficacy continues through the 6-to-12-month window, supporting treatment beyond early infancy when needed. [7] Pope and colleagues' randomised trial showed that nadolol is non-inferior to propranolol with a once-daily dosing advantage. [6]
The propranolol side effects the candidate must name are hypoglycaemia (especially when feeding falls during an intercurrent illness), bradycardia, hypotension, bronchospasm, sleep disturbance and gastrointestinal upset. [5] Mitigate them by feeding around dose times, monitoring heart rate and blood pressure, avoiding propranolol in infants with clinically significant bradycardia, heart block or reactive airway disease, and giving parents clear "sick-day" instructions to hold the dose and seek review when the infant is not feeding. [5]
For the port-wine stain, the definitive treatment is pulssed-dye laser, ideally begun in infancy when the stain is lighter and the lesion smaller, accepting that it improves rather than erases the mark. [9] When the stain is in the V1 distribution, the more urgent task is surveillance: gadolinium brain MRI for leptomeningeal angiomatosis, ophthalmology for glaucoma (which can present in infancy), and anticonvulsant prophylaxis or treatment for the seizures that often declare in the first two years of Sturge–Weber syndrome. [9]
For the giant congenital melanocytic naevus, management is multidisciplinary and individualised: neurological surveillance for neurocutaneous melanocytosis (baseline brain and spine MRI, monitoring for signs of raised intracranial pressure or neurological decline), lifelong dermatological surveillance for melanoma, psychological support for the child and family, and a decision about surgical excision, tissue expansion, or watchful waiting based on site, size and the family's preference. [13]
For dermal melanocytosis, management is reassurance and documentation: measure and photograph the lesion at birth so that any later concern about a bruise can be resolved by comparison, and review persistent or atypical spots. [10] For the midline lumbosacral lesion, refer early for spinal ultrasound and, when abnormal, neurosurgical review. [4]
The haemangioma decision in one pass
1 · Assess
Site, size, segmental pattern, periocular/airway involvement, ulceration, number of lesions
2 · Triage
Observe if small/uncomplicated; escalate if large/facial/periocular/airway/segmental or ≥5 lesions
3 · Treat
Topical timolol for small superficial lesions; oral propranolol for higher-risk lesions
4 · Screen
PHACE work-up for large segmental facial; liver ultrasound + thyroid if ≥5 lesions
5 · Follow-up
Photograph through proliferation, monitor propranolol side effects, wean at 12–18 months, refer residual deformity
Specific Subtypes & Scenarios
Each high-yield scenario rewards a candidate who can give the decision, the work-up, and the counselling in one breath. [4]
The small uncomplicated haemangioma on the trunk or limb is observed with serial photographs. The family is counselled that the lesion will grow for some months and then shrink over years, usually leaving little trace, and that treatment is rarely needed. [4]
The periocular haemangioma is the scenario that tests whether a candidate understands urgency. A haemangioma of the upper eyelid threatens vision by causing deprivation amblyopia (blocking the visual axis) and astigmatism (distorting the globe). Refer to ophthalmology within days, start oral propranolol early, and review refractive status repeatedly through proliferation. [4]
The segmental facial haemangioma over 5 cm carries a risk of PHACE syndrome — posterior fossa brain malformations (classically the Dandy–Walker complex), the segmental Haemangioma, Arterial cerebrovascular anomalies, Cardiac coarctation or aortic arch anomaly, and Eye anomalies. The work-up is brain MRI/MRA, echocardiogram and aortic-arch imaging, and ophthalmology. [8]
Five or more cutaneous haemangiomas trigger an abdominal ultrasound for hepatic haemangioma and a thyroid panel for consumptive hypothyroidism; large hepatic lesions can also cause high-output cardiac failure. [4]
The V1 port-wine stain is the Sturge–Weber scenario. Refer for gadolinium brain MRI, ophthalmology for glaucoma, and begin pulsed-dye laser. Counsel the family about the risk of seizures, the need for developmental surveillance, and that a normal early MRI does not eliminate later risk. [9]
The midline lumbosacral lesion — a dimple more than 2.5 cm from the anal verge and/or larger than 5 mm with other markers, a lipoma, a hair tuft, a skin tag or a deviated gluteal cleft — is a sentinel for occult spinal dysraphism and tethered cord. Refer for spinal ultrasound before six months, and neurosurgery if abnormal. [4]
Erythema toxicum versus suspected infection. A well term infant with a classic migratory pustular rash sparing the palms and soles needs no tests. An unwell or premature infant, or a pustule with surrounding induration or a clustered or haemorrhagic morphology, warrants a Wright stain, culture and HSV PCR, and empirical treatment of the suspected organism while results are awaited. [12]
The neonatal blister. A neonate with widespread blistering or erosion needs same-day paediatric dermatology. Take swabs for HSV PCR and bacterial culture, consider staphylococcal scalded skin syndrome, herpes simplex, epidermolysis bullosa and incontinentia pigmenti, and treat empirically for sepsis and herpes while results are awaited. [11]
Complications & Pitfalls
The complications fall into those of the lesions themselves and those of their treatment, and both are examinable. [4]
Untreated or higher-risk infantile haemangiomas cause ulceration (most often the lip, neck fold or perineum), bleeding, secondary infection, scarring and disfigurement. Periocular lesions cause amblyopia and astigmatism; airway lesions cause stridor and obstruction; hepatic lesions can cause high-output cardiac failure and consumptive hypothyroidism. The Kasabach–Merritt phenomenon is the most dangerous complication of all, a consumptive coagulopathy behind a kaposiform haemangioendothelioma or tufted angioma. [4]
The propranolol complications are the ones a candidate must recite unprompted. Hypoglycaemia is the most feared: propranolol masks the adrenergic warning signs, so a hypoglycaemic infant on propranolol may simply be quiet or jittery, and the risk is highest during an intercurrent illness with reduced feeding. [5] Give parents explicit sick-day rules: hold the dose if the infant is off feeds, check in with the team, and seek review. Bradycardia, hypotension, bronchospasm (a relative contraindication in reactive airway disease), sleep disturbance and gastrointestinal upset are the other named effects. [5]
The diagnostic pitfalls are the ones that lose marks and harm children. Misreading a port-wine stain of the forehead as a salmon patch and missing Sturge–Weber surveillance is a recurring error. [9] Dismissing a midline lumbosacral lesion and missing a tethered cord is another. [4] Mistaking a Mongolian spot for a bruise and raising a safeguarding referral in error — or, in reverse, dismissing a real bruise on a non-mobile infant as a birthmark — is the third. Document every birthmark's size and site at the newborn check so that later comparisons are possible. [10]
The fourth pitfall is treating the benign majority: applying topical steroids to erythema toxicum, prescribing antibiotics for milia, or laser-treating a salmon patch that would have faded on its own. [11]
The long-term risks belong to the syndromic lesions: the seizure, stroke-like episodes, glaucoma and developmental risk of Sturge–Weber; the neurocutaneous melanocytosis and melanoma risk of the giant congenital melanocytic naevus; and the cerebrovascular and cardiac risk of PHACE. [8] [9] [13]
Prognosis & Disposition
Prognosis is reassuring for the great majority and is determined, for the minority, by the syndromic lesion. [4]
The benign transient lesions all resolve fully without sequelae: erythema toxicum and milia within days to weeks, transient neonatal pustular melanosis over weeks to months (leaving pigmentation that fades), and dermal melanocytosis over the first years in most populations. [11]
The infantile haemangioma follows its famous life cycle: proliferation to roughly 9 months, plateau to about 12 months, then involution at roughly 10 per cent per year of the remaining lesion — about half involuted by age 5 and about 90 per cent by age 9. [4] The residual skin may show telangiectasia, pallor, fibrofatty tissue or redundant skin that may need later surgical or laser correction. [4]
The port-wine stain is lifelong; pulsed-dye laser lightens but rarely erases it, and the lesion thickens and develops vascular blebs in adult life. The prognosis of Sturge–Weber syndrome is shaped by the leptomeningeal involvement — the frequency and control of seizures, the risk of stroke-like episodes and glaucoma, and the developmental trajectory — which is why surveillance begins in infancy. [9]
The giant congenital melanocytic naevus carries a lifetime melanoma risk often quoted around 2 to 5 per cent, and a risk of neurocutaneous melanocytosis that is highest with large axial naevi and many satellite lesions. [13] Neurological and dermatological surveillance continue through childhood. [13]
Disposition is straightforward: the small uncomplicated haemangioma and all transient lesions stay in primary care with parental reassurance and a safety-net; any syndromic or serious lesion goes to a multidisciplinary paediatric service (dermatology, ophthalmology, ENT, cardiology, neurology and neurosurgery as the lesion dictates). [4]
Special Populations
Care changes with the infant and the family, and the candidate who shows that awareness earns marks at the long case and the communication station. [4]
In the preterm infant, the skin is thinner, more vulnerable to injury and to temperature and fluid loss, and erythema toxicum may appear later or look atypical. Preterm and low-birth-weight infants also have a higher incidence of infantile haemangioma, and propranolol is initiated with closer monitoring of heart rate, blood pressure, feeding and glucose in this group. [3]
Infants with complex syndromic lesions — PHACE, Sturge–Weber, giant congenital melanocytic naevus — are managed in a multidisciplinary team across dermatology, neurology, ophthalmology, cardiology, neurosurgery and psychology, and the paediatrician often coordinates the surveillance and the interface with school and developmental services. [8] [9]
Cultural and safeguarding considerations matter here. Dermal melanocytosis is near-universal in many Asian, Polynesian, Indigenous Australian and African populations, and misreading a typical spot as a bruise is a known cause of unnecessary safeguarding referrals. [10] The reverse is also true: a bruise on a non-mobile infant must not be dismissed as a birthmark. The safeguarding interface turns on careful documentation of size, site, evolution and context, and on knowing the typical sites and appearance of dermal melanocytosis. [10]
Socioeconomic and rural-remote access shapes who reaches paediatric dermatology, laser and propranolol services, and who gets the multidisciplinary care a syndromic lesion needs. Telehealth dermatology, regional propranolol initiation under shared-care protocols, and clear referral pathways reduce the gap, and the candidate should name them. [4]
Parent counselling is the thread that runs through every lesion. Use the child's name, show the lesion, explain what it is and what it will do, give written or photographic information, and arrange a clear follow-up. The reassurance given to the family of a baby with erythema toxicum is as much the job as the propranolol prescription is for a baby with a periocular haemangioma. [11]
Evidence, Guidelines & Regional Differences
The evidence base for this topic is unusually clinician-shaping, and a candidate should know the landmark papers and how they changed practice. [2]
The propranolol revolution began with the 2008 observation by Léauté-Labrèze and colleagues, who reported regression of severe haemangiomas in infants given propranolol for cardiac indications. [2] That single observation transformed infantile haemangioma from a disease of watchful waiting, corticosteroids and surgery into one of safe, effective β-blockade. The AAP clinical report of Darrow and colleagues (2015) set out the diagnosis, the indications for treatment, and the dose and monitoring of propranolol, and is the standard reference for North American and much international practice. [4] The consensus conference of Drolet and colleagues (2013) codified propranolol initiation, dose escalation and monitoring, including the contraindications and the sick-day counselling. [5]
On the Sturge–Weber side, the 2013 paper by Shirley and colleagues established that a postzygotic GNAQ mutation underlies both the port-wine stain and the syndrome, reframing surveillance around the dermatomal distribution of the stain and seeding molecular-target research. [9] The PHACE consensus of Metry and colleagues (2009) set the diagnostic criteria for PHACE syndrome and remains the reference for work-up. [8]
The evidence is weaker in several places worth naming. The optimal timing of pulsed-dye laser for port-wine stain, the role of prophylactic excision of giant congenital melanocytic naevi, the ideal duration of propranolol treatment, and the place of topical β-blockers in deeper haemangiomas remain debated — a candidate who acknowledges the uncertainty while giving a defensible plan is reading the field correctly. [4] [13]
Exam Pearls
The high-yield facts a candidate should hold ready. [4]
PHACE
The Sturge–Weber rule the candidate must state plainly: the risk of leptomeningeal angiomatosis and glaucoma is concentrated when the port-wine stain involves the forehead or upper eyelid (the V1 dermatome); a port-wine stain confined to the cheek carries a much lower risk. [9]
The propranolol facts to recite unprompted: oral 2 to 3 mg/kg/day in divided doses, started at 2 and titrated to 3 mg/kg/day, continued through proliferation to roughly 12 to 18 months, weaned rather than stopped, with heart-rate and blood-pressure monitoring and explicit sick-day counselling for the risk of silent hypoglycaemia. [4] [5]
The referral rules to hold ready: five or more cutaneous haemangiomas trigger a liver ultrasound and thyroid panel; a midline lumbosacral lesion triggers spinal ultrasound before six months; a facial segmental haemangioma over 5 cm triggers a PHACE work-up; a V1 port-wine stain triggers brain MRI and ophthalmology. [8] [9]
The pustular-rash discriminator: erythema toxicum spares the palms and soles and shows eosinophils on a Wright stain; transient neonatal pustular melanosis can affect the palms and soles, leaves pigmentation, and shows neutrophils. [12] Milia are 1 mm white epidermoid cysts on the nose; sebaceous hyperplasia is yellow pinpoint papules on the nose; both are maternal-androgen driven and self-limiting. [11] Harlequin colour change is a transient, sharp midline colour split in the first week, benign and self-limiting. [11]
Finally, the giant congenital melanocytic naevus: a projected adult size over 20 cm defines the high-risk category, with satellite lesions and an axial site raising the risk of neurocutaneous melanocytosis and melanoma, both of which change the surveillance plan. [13]
References
- [1]Mulliken JB, Glowacki J Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plastic and Reconstructive Surgery, 1982.PMID 7063565
- [2]Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A Propranolol for severe hemangiomas of infancy. New England Journal of Medicine, 2008.PMID 18550886
- [3]Haggstrom AN, Drolet BA, Baselga E, et al (Hemangioma Investigator Group) Prospective study of infantile haemangiomas: demographic, prenatal, and perinatal characteristics. Journal of Pediatrics, 2007.PMID 17307549
- [4]Darrow DH, Greene AK, Mancini AJ, Nopper AJ (American Academy of Pediatrics) Diagnosis and management of infantile hemangioma. Pediatrics, 2015.PMID 26416931
- [5]Drolet BA, Frommelt PC, Chamlin SL, et al Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics, 2013.PMID 23266923
- [6]Pope E, Chakkittakandiyil A, Lara-Corrales I, et al Noninferiority and safety of nadolol vs propranolol in infants with infantile hemangioma: a randomized controlled trial. JAMA Pediatrics, 2022.PMID 34747977
- [7]Baselga E, Dembowska-Bagińska B, Przewratil P, et al Efficacy of propranolol between 6 and 12 months of age in high-risk infantile hemangioma. Pediatrics, 2018.PMID 30082451
- [8]Metry DW, Haggstrom AN, Drolet BA, et al Consensus statement on diagnostic criteria for PHACE syndrome. Pediatrics, 2009.PMID 19858157
- [9]Shirley MD, Tang H, Gallione CJ, et al Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. New England Journal of Medicine, 2013.PMID 23656586
- [10]Leung AK, Kao CP, Lee TK Persistent Mongolian spots in Chinese adults. International Journal of Dermatology, 2005.PMID 15663659
- [11]Patrizi A, Raone B, Neri I, D'Acunto C Advances in pharmacotherapeutic management of common skin diseases in neonates and infants. Expert Opinion on Pharmacotherapy, 2017.PMID 28429969
- [12]Chadha A, Yau E, Alikhan A Common neonatal rashes. Pediatric Annals, 2019.PMID 30653638
- [13]Krengel S, Scope A, Dusza SW, Vonthein R, Marghoob AA New recommendations for the categorization of cutaneous features of congenital melanocytic nevi. Journal of the American Academy of Dermatology, 2013.PMID 22982004