Paeds · gastroenterology-hepatology-and-nutrition
Coeliac disease
Also known as Coeliac disease · Celiac disease · Gluten-sensitive enteropathy · Gluten enteropathy · Coeliac sprue · Non-tropical sprue
Fellowship guide to coeliac disease in children: the immune-mediated small-bowel enteropathy triggered by gluten in genetically susceptible people, the classic and non-classic presentations from the wasted toddler with a distended abdomen to the adolescent with short stature or refractory iron-deficiency anaemia, the HLA-DQ2 and DQ8 genetic background, the immunoglobulin A anti-tissue-transglutaminase-first serological workup with total immunoglobulin A to exclude deficiency, the Marsh histology, the ESPGHAN 2020 no-biopsy pathway requiring a titre at or above ten times the upper limit of normal plus positive endomysial antibody on a separate sample, the gluten-on-board rule, and the lifelong strict gluten-free diet with dietitian support and monitoring.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Coeliac disease is a permanent, immune-mediated enteropathy of the small intestine triggered by gluten in genetically susceptible people, and it is one of the commonest chronic diseases of childhood once it is actively looked for. The whole task of the clinician is to recognise the child whose faltering growth, refractory anaemia, abdominal distension or persistent diarrhoea is the face of untreated coeliac disease, confirm the diagnosis with serology taken while the child is still eating gluten, and settle the family onto a strict lifelong gluten-free diet. [1]
Gluten is the alcohol-soluble protein fraction of wheat, barley and rye, and its gliadin component drives the immune injury. In a susceptible child, eaten gluten provokes an autoimmune response in the lamina propria that flattens the small-bowel mucosa, so that the tall, finger-like villi become ridges, the crypts lengthen, and the absorptive surface is lost. The result is malabsorption of iron, folate, calcium and fat-soluble vitamins, and the many clinical faces of the disease flow from that single lesion. [3]
The modern definition is broad. The Oslo definitions frame coeliac disease as a gluten-induced enteropathy with a variable clinical presentation, so the wasted toddler with diarrhoea is only the classic tip of an iceberg that also includes the school-age child with isolated short stature, the adolescent with iron deficiency that will not correct with oral iron, and the asymptomatic sibling picked up by screening. The unifying feature is a gluten-dependent small-bowel enteropathy that resolves on a gluten-free diet. [3]
Classification
The most useful clinical way to hold coeliac disease is by the face it shows, because the classic wasted toddler is now a minority of cases. The Oslo definitions separate the presentations into classic disease, where the malabsorption dominates with chronic diarrhoea, steatorrhoea, failure to thrive and a distended abdomen, and non-classic disease, where a single extraintestinal feature such as iron-deficiency anaemia, short stature, delayed puberty, osteoporosis or raised liver enzymes is the only clue. Recognising the non-classic face is what prevents the missed adolescent diagnosis. [3]

A second axis is whether the disease is symptomatic or detected by screening. Symptomatic coeliac disease presents to the clinician with complaints; silent coeliac disease has no symptoms but positive serology and villous atrophy, found when first-degree relatives, children with type 1 diabetes, Down syndrome, Turner syndrome, selective immunoglobulin A deficiency or autoimmune thyroid disease are screened; and potential coeliac disease has the antibodies but a normal small-bowel biopsy, carrying a risk of future evolution into active disease. These screened groups matter because they are the reservoir of unrecognised coeliac disease in the community. [1] [3]
A third axis is the severity of the mucosal lesion, graded by the Marsh classification. Marsh 1 is an infiltrive lesion with raised intraepithelial lymphocytes but normal villi; Marsh 2 adds crypt hyperplasia; and Marsh 3 is the hyperplastic-atrophic lesion with villous atrophy, split into Marsh 3a for partial, 3b for subtotal and 3c for total villous atrophy. The Marsh grade does not always track with the severity of symptoms, but it is the histological confirmation of the enteropathy when a biopsy is needed. [1]
Epidemiology & Risk Factors
Coeliac disease is common wherever gluten is eaten and the genes are present. A systematic review of global data put the seroprevalence at around one point four per cent and the biopsy-confirmed prevalence at around zero point seven per cent, so roughly one in a hundred children is affected, with a slight female predominance. The true burden is higher than the diagnosed burden, because most coeliac disease sits below the clinical waterline and is only found when it is looked for. [6]
The single strongest risk factor is the genetic background. Around ninety to ninety-five per cent of people with coeliac disease carry the HLA-DQ2 heterodimer, and almost all of the remainder carry HLA-DQ8, so the absence of both makes coeliac disease very unlikely and gives human leukocyte antigen typing its high negative predictive value. First-degree relatives carry roughly a one in ten lifetime risk, monozygotic twins show around seventy per cent concordance, and the genes are necessary but not sufficient, since most gene carriers never develop the disease. [1] [6]
Autoimmune and chromosomal disorders raise the risk and define the groups who should be screened. Type 1 diabetes carries the clearest association, with roughly three to eight per cent of children with diabetes also having coeliac disease, and Down syndrome carries an even higher relative risk. Selective immunoglobulin A deficiency, Turner syndrome, Williams syndrome and autoimmune thyroiditis all increase the likelihood, so the at-risk child is part of the routine screening population in most paediatric gastroenterology services. [4]
Pathophysiology
The lesion is built from a gene, a food protein and an enzyme. In a child who carries HLA-DQ2 or HLA-DQ8, eaten gluten reaches the lamina propria, where the enzyme tissue transglutaminase deamidates specific glutamine residues in the gliadin peptide. The deamidated peptide binds the disease-associated HLA-DQ2 or DQ8 molecule on antigen-presenting cells far more tightly than native gliadin, so it is presented powerfully to gluten-specific CD4-positive T cells, which then drive a T-helper-1 inflammatory cascade and a B-cell response that produces the autoantibodies. [1] [4]
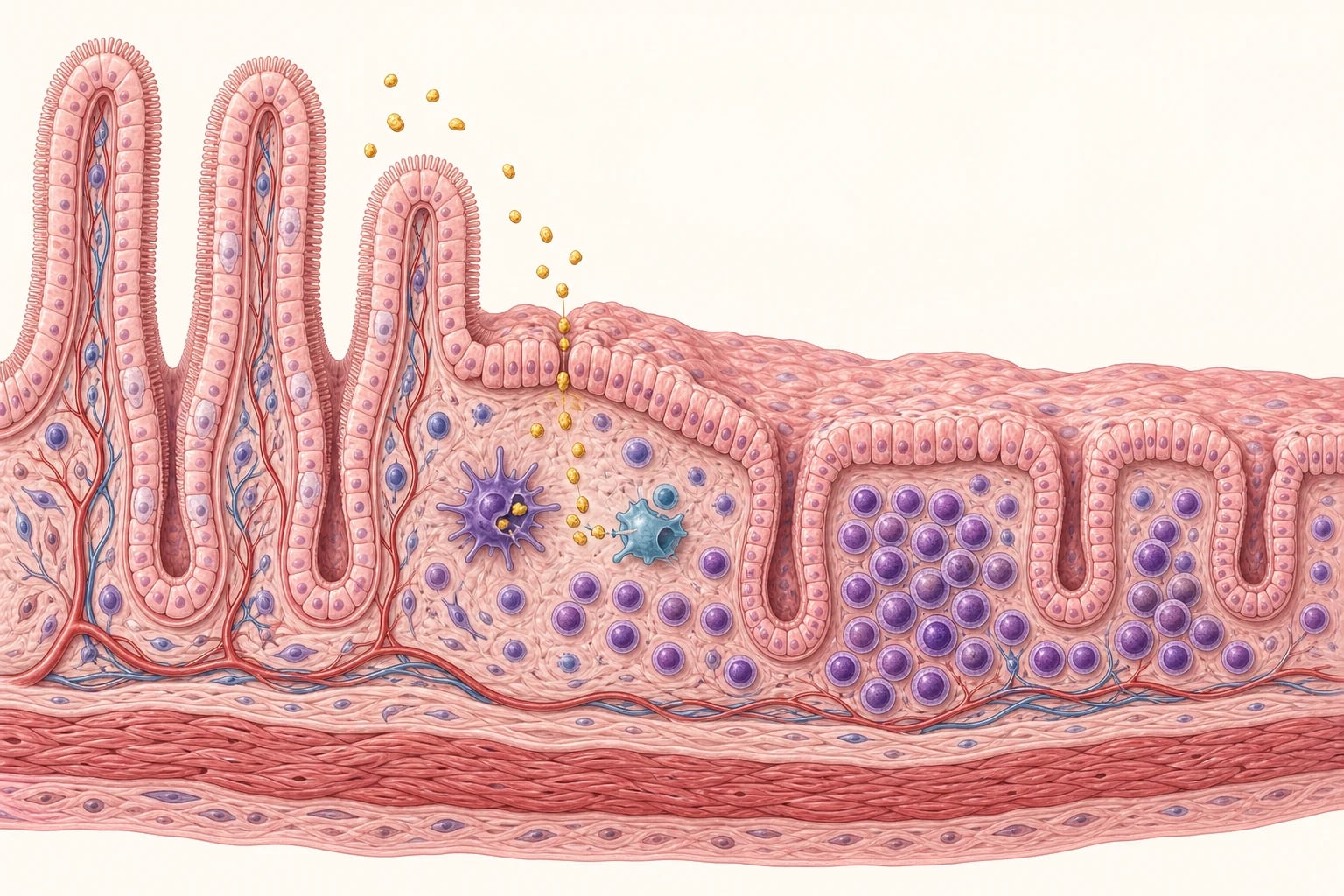
The same tissue transglutaminase is the target of the autoantibodies that define the serology. The inflammatory cytokines, led by interferon-gamma, drive intraepithelial lymphocyte infiltration, crypt hyperplasia and villous atrophy, so the absorptive surface shrinks and brush-border enzymes fall. Loss of the absorptive surface and of disaccharidase activity then produces the malabsorption of iron, folate, calcium and fat-soluble vitamins, and secondary lactose intolerance from lactase deficiency can layer on top of the primary disease. [4]
The lesion is entirely gluten-dependent, which is the principle that underpins both diagnosis and treatment. Remove the gluten and the immune stimulus disappears, the antibodies fall over weeks to months, and the mucosa heals over months to years; reintroduce the gluten and the whole process restarts. This reversibility is why the serology must be taken while the child is still eating gluten, why a gluten challenge is needed if the diet has already been started, and why the only treatment is a lifelong gluten-free diet. [1] [11]
Clinical Presentation
The classic face is the toddler, seen less often now than a generation ago. After gluten is introduced at weaning, the young child develops chronic or intermittent diarrhoea, a distended abdomen, wasted buttocks and thighs, irritability, anorexia and vomiting, and falls away from the growth centiles for weight and length. The picture of a miserable, pot-bellied, thin-limbed toddler with failure to thrive after starting cereals is the classic presentation, and although it remains important, it is now the minority way the disease declares itself. [4] [1]
The non-classic face dominates in the school-age child and adolescent, and it is the one most often missed. Refractory iron-deficiency anaemia is the commonest single clue, because iron is absorbed at the duodenal tip where the atrophy is worst; short stature and delayed puberty, chronic abdominal pain and bloating, chronic constipation, recurrent aphthous mouth ulcers, dental enamel defects of the permanent teeth, arthritis, unexplained raised transaminases, osteoporosis and fractures, epilepsy with cerebral calcifications, and the itchy blistering rash of dermatitis herpetiformis are all recognised monosymptomatic presentations. [4] [11]
The silent face is found by screening. The child with type 1 diabetes, Down syndrome, selective immunoglobulin A deficiency or an affected first-degree relative may have no gut complaint at all yet carry positive serology and villous atrophy, and it is the screening programme that brings them to light. Because untreated coeliac disease can quietly impair growth, bone density and glycaemic control in the child with diabetes, these screened children are still offered treatment even when they feel well. [1] [6]
Differential Diagnosis
The differential depends on which face the disease shows. For the classic toddler with diarrhoea and failure to thrive, the competitors are cow's milk protein enteropathy, post-enteritis enteropathy with secondary lactose malabsorption, cystic fibrosis with pancreatic insufficiency, chronic giardiasis, toddler's diarrhoea in the thriving child, and the rare congenital enteropathies. The pale, bulky, greasy steatorrhoeic stool points towards fat malabsorption and prompts faecal elastase and a sweat test alongside the coeliac serology. [4]
For the older child with refractory iron-deficiency anaemia or short stature, the differential is the whole list of causes of each, and coeliac disease is the enteropathy that links them. Dietary iron deficiency, menorrhagia in the adolescent girl, helicobacter gastritis, inflammatory bowel disease, chronic blood loss from a Meckel diverticulum or polyp, and endocrine causes of short stature such as growth hormone deficiency and hypothyroidism all belong on the list, and coeliac serology is the single test that screens the gut cheaply and effectively in all of them. [11] [4]
For the child with chronic abdominal pain and bloating, the functional disorders dominate once the red flags are excluded. Irritable bowel syndrome, functional abdominal pain, lactose intolerance, small intestinal bacterial overgrowth, and chronic constipation all mimic the discomfort of coeliac disease, and the value of the serology is that it confidently removes the enteropathy from the differential before a functional label is attached. The must-not-miss diagnosis is coeliac disease itself, missed because the presentation is monosymptomatic and the gluten-on-board rule is broken by a premature diet. [4] [1]
Monosymptomatic presentations of coeliac disease — 'GROWTH-IRIS'
Clinical & Bedside Assessment
Assessment begins, as in all of paediatrics, with the growth chart. Plot the weight, the height and the body mass index, read the trend across the centiles, and look for the fall that signals an organic enteropathy, because coeliac disease can present as isolated short stature with a completely normal bowel habit. Delayed puberty, a low body mass index, pallor from anaemia and the wasted buttocks with a distended abdomen of the classic case are the physical stigmata to seek. [4] [11]
The history then hunts the face of the disease and the company it keeps. Ask about the stool pattern, the abdominal pain and bloating, the appetite, the mouth ulcers, the dental enamel, the joint pains, the rash, the fatigue and the pubertal stage. Ask specifically about type 1 diabetes, thyroid disease, Down syndrome and selective immunoglobulin A deficiency in the child, and about coeliac disease, autoimmune disease and consanguinity in the family. Record the age at which gluten was introduced and confirm that the child is still eating a normal gluten-containing diet. [1] [4]
Examination is often normal, which is the trap. Look for pallor, short stature, a distended abdomen, wasted buttocks and proximal muscles, aphthous ulcers, dental enamel hypoplasia, finger clubbing, a rash over the elbows and knees consistent with dermatitis herpetiformis, and the stigmata of Down syndrome or the injection sites of type 1 diabetes. The combination of a normal abdominal examination with a faltering growth chart or a refractory anaemia is exactly the picture that demands coeliac serology. [11] [4]
Investigations
Investigation is built on serology taken while the child is still eating gluten. The first test is immunoglobulin A anti-tissue-transglutaminase antibody, the most accurate single screen, and it must always be paired with a total serum immunoglobulin A to exclude selective immunoglobulin A deficiency, which occurs in roughly two to three per cent of children with coeliac disease and would render the test falsely negative. The anti-tissue-transglutaminase test carries a sensitivity of around ninety-three per cent and a specificity of ninety-seven to ninety-nine per cent, and endomysial antibody is comparably accurate but more observer-dependent, so it is used as the confirmatory second test rather than the screen. [5] [9]
The ESPGHAN 2020 guideline defines two routes to the diagnosis, and the no-biopsy pathway is the one examiners probe. A symptomatic child whose immunoglobulin A anti-tissue-transglutaminase titre is at or above ten times the upper limit of normal, with a positive endomysial antibody on a separate serum sample and a normal total immunoglobulin A, can be diagnosed with coeliac disease without a duodenal biopsy. Human leukocyte antigen typing and the symptom profile are no longer required for the no-biopsy route in the 2020 revision, which broadened access to the pathway by dropping the earlier HLA condition. [1] [8]
When the no-biopsy criteria are not met, the standard remains the duodenal biopsy with multiple samples, including from the duodenal bulb. The histology shows villous atrophy with crypt hyperplasia and increased intraepithelial lymphocytes, graded by the Marsh classification, and it is required when the titre is below ten times the upper limit of normal, when the serology and the picture are discordant, or when selective immunoglobulin A deficiency forces the use of less specific immunoglobulin G-based tests. Human leukocyte antigen typing for DQ2 and DQ8 is reserved for ruling the disease out when the serology and histology are inconclusive, because its negative predictive value is high and its positive predictive value is low. [1] [2]
Supporting blood tests stage the malabsorption and the deficiencies. A full blood count and film show the microcytic iron-deficiency anaemia, ferritin and folate are low, vitamin D and other fat-soluble vitamins may be deficient, calcium and phosphate can be low with a raised parathyroid hormone, and liver transaminases may be mildly raised. Coagulation, thyroid function and a bone profile complete the staging, and the deficiencies guide the early nutritional correction that runs alongside the diet. Human immunodeficiency and tissue typing are not part of the routine workup. [11] [4]
Management — Resuscitation

Most coeliac disease is managed electively in clinic, but a small number of children present acutely and need stabilisation first. The rare coeliac crisis presents with profuse diarrhoea, severe dehydration, hypokalaemia, metabolic acidosis and hypoproteinaemia in a deeply malnourished child, and it is a medical emergency. Assess the airway, breathing and circulation, correct the circulating volume and the fluid and electrolyte deficits, replace potassium carefully because chronic diarrhoea depletes total-body potassium more than the plasma level suggests, and begin careful nutritional support, with a short course of corticosteroids reserved for the severe crisis that does not settle with resuscitation alone. [1] [11]
Severe iron-deficiency anaemia with haemodynamic compromise is the other acute presentation that may need action before the diet is started. A profoundly anaemic, tachycardic child may need a cautious blood transfusion, while the more common moderate anaemia is corrected with oral iron once the diagnosis is made. The undernourished child with faltering growth needs structured nutritional rehabilitation with attention to refeeding, because rapid reintroduction of calories in a malnourished child can precipitate the refeeding syndrome with hypophosphataemia, hypokalaemia and thiamine deficiency. [11] [12]
Oral iron for coeliac-associated iron-deficiency anaemia
Loading dose
Elemental iron 3 mg/kg once daily for infants and young children, increased to a divided dose up to 6 mg/kg per day if tolerated
Maintenance dose
Ferrous sulfate 200 mg once or twice daily for older children and adolescents, continued for around three months after the anaemia resolves and then reassessed on the gluten-free diet
Immediate management of the acutely unwell child with coeliac crisis or severe deficiency
Assess ABC, hydration and nutritional status; send electrolytes, glucose, a blood gas, full blood count and albumin
Correct dehydration and shock with intravenous fluids, then move to maintenance with careful electrolyte replacement
Replace potassium cautiously, because total-body depletion exceeds the plasma level
Start careful nutritional rehabilitation and watch for refeeding syndrome, with thiamine, phosphate and potassium monitoring
Correct severe iron-deficiency anaemia; transfuse cautiously only if haemodynamically unstable
Once stable, proceed to the gluten-containing-diet serology and the formal diagnostic pathway
Management — Definitive & Stepwise
The definitive treatment is a strict, lifelong gluten-free diet, and there is no role for medication in routine management. The diet excludes all wheat, barley and rye, which means no ordinary bread, pasta, biscuits, cakes, breakfast cereals or beer, and a close reading of labels for hidden gluten in sauces, processed foods and thickeners. Pure, uncontaminated oats are tolerated by most children with coeliac disease, but oats must be labelled gluten-free to avoid cross-contamination from wheat in the supply chain, and a few children also react to oat proteins. [11] [1]
The diet is delivered with dietitian support and monitored by the clinical and serological response. Symptoms usually settle within weeks, growth catches up over months, and the immunoglobulin A anti-tissue-transglutaminase titre falls progressively, typically normalising within six to twelve months on a strict diet, so a persistently raised titre is a sensitive marker of ongoing gluten exposure. Annual review checks the weight and height, the symptoms, the antibody titre, the full blood count and ferritin, the bone profile and vitamin D, and the adherence, and a bone density scan is considered in the adolescent with persistently poor intake or delayed puberty. [11] [4]
Correcting the deficiencies and screening the family complete the management. Iron, folate, calcium and vitamin D are replaced as guided by the baseline bloods, and the autoimmune associations mean that thyroid function, type 1 diabetes screening and pubertal staging are kept under surveillance. First-degree relatives are offered serological screening, and the child is supported through adolescence with a planned transition to adult coeliac services, because adherence often falls in the teenage years just as the long-term risks of untreated disease begin to accumulate. [11] [12]
The ESPGHAN 2020 no-biopsy pathway — the detail examiners probe
The 2020 ESPGHAN guideline allows coeliac disease to be diagnosed without a duodenal biopsy in a symptomatic child when the immunoglobulin A anti-tissue-transglutaminase titre is at or above ten times the upper limit of normal, a second separate blood sample shows positive endomysial antibody, and the total immunoglobulin A is normal. The 2020 revision dropped the earlier requirement for a compatible human leukocyte antigen type and specific symptoms, broadening access to the no-biopsy route. When the criteria are not met, the standard remains duodenal biopsy with multiple biopsies including the bulb, showing villous atrophy, crypt hyperplasia and raised intraepithelial lymphocytes. Every test must be taken while the child is still eating gluten. [1] [8]
Specific Subtypes & Scenarios
The screened child with type 1 diabetes is the scenario that tests breadth of practice. Roughly three to eight per cent of children with type 1 diabetes also have coeliac disease, so ESPGHAN recommends serological screening at diagnosis and periodically thereafter, using immunoglobulin A anti-tissue-transglutaminase with a total immunoglobulin A. A positive screen in an asymptomatic child still warrants treatment, because untreated coeliac disease can impair growth, reduce bone density and destabilise glycaemic control, and the gluten-free diet must be balanced against the carbohydrate-counting needs of diabetes care with a specialist dietitian. [1] [11]
The child with selective immunoglobulin A deficiency is the serology trap. Because the standard serology is immunoglobulin A-based, a child who is both coeliac and immunoglobulin A-deficient returns a falsely negative anti-tissue-transglutaminase, which is why a total immunoglobulin A must accompany every screen. When deficiency is found, diagnosis relies on immunoglobulin G-based tests such as anti-deamidated gliadin peptide immunoglobulin G and the immunoglobulin G anti-tissue-transglutaminase, supported by human leukocyte antigen typing and, where needed, duodenal biopsy. [1] [5]
The child already on a gluten-free diet is the diagnostic trap. Families and complementary practitioners often start a gluten-free diet empirically, and because the antibodies fall and the mucosa heals within weeks to months, the serology and biopsy then become uninterpretable. The diagnosis can only be confirmed by a formal gluten challenge, which means reintroducing a gluten-containing diet for several weeks and repeating the serology and biopsy, a process that is unpleasant, symptom-laden and best avoided by testing before the diet is ever started. [1] [4]
Across Australia, New Zealand and the United Kingdom, coeliac disease is worked up in primary care or general paediatrics with immunoglobulin A anti-tissue-transglutaminase plus total immunoglobulin A on a gluten-containing diet, and the no-biopsy pathway is applied by general paediatricians and gastroenterologists alike for children who meet the 2020 criteria. The cost and availability of gluten-free products hit families unevenly, and in some jurisdictions subsidised gluten-free staples are available on prescription or through coeliac societies. In many low- and middle-income settings the disease is underdiagnosed, the diagnostic antibody tests and the gluten-free foods are scarce or costly, and the burden of undiagnosed malabsorption is carried by remote, Indigenous and migrant communities, who need culturally safe shared-care pathways and access to affordable gluten-free alternatives. [6] [11]
Complications & Pitfalls
The complications of untreated or poorly controlled coeliac disease flow from chronic malabsorption and chronic inflammation. Persistent failure to thrive and short stature, delayed puberty, iron-deficiency and folate-deficiency anaemia, osteopenia and osteoporosis with fractures, and unexplained raised transaminases are the everyday consequences of an uncontrolled diet. A large cohort linked the degree of small-intestinal villous atrophy to a small but real increase in mortality, underlining that the disease is not trivial even when the child looks well. [12] [11]
The serious long-term complications are malignancy and refractory disease, though both are rare in childhood. Untreated coeliac disease carries an increased lifetime risk of enteropathy-associated T-cell lymphoma and small-bowel adenocarcinoma, risks that fall towards the background with a strict gluten-free diet, and refractory coeliac disease, defined by persistent symptoms and villous atrophy despite a strict diet for more than twelve months, is uncommon in children but demands specialist reassessment for lymphoma and other causes of non-responsive enteropathy. Dermatitis herpetiformis is the intensely itchy blistering rash over elbows, knees and buttocks, controlled by a gluten-free diet with dapsone for the rash in the early phase. [12] [11]
The diagnostic pitfalls are the failures the examiner rewards for naming. The first is testing serology after gluten has been withdrawn, which normalises the antibodies and forces a difficult gluten challenge. The second is omitting the total immunoglobulin A and missing selective immunoglobulin A deficiency, so a coeliac child returns a falsely negative screen. The third is over-diagnosing on a single low-titre positive screen without confirmation or biopsy, or labelling a non-coeliac gluten sensitivity as coeliac disease when the serology and biopsy are normal. [1] [4]
Prognosis & Disposition
The prognosis for the child who adheres to a strict gluten-free diet is excellent. Symptoms resolve within weeks, growth and puberty catch up over months, the antibody titre falls to normal within six to twelve months, and the mucosa heals over one to two years. The long-term risks of untreated disease, including osteoporosis, refractory anaemia, infertility and the small excess of lymphoma and small-bowel malignancy, fall towards the background on a strict diet, so adherence is the single most important determinant of outcome. [11] [12]
The prognosis is more guarded for the poorly adherent teenager and for refractory disease. Adherence often declines in adolescence just as independence in food choice grows, so structured transition to adult coeliac services, repeat serology and dietitian contact are the safeguards. Refractory coeliac disease, enteropathy-associated T-cell lymphoma and small-bowel adenocarcinoma are rare in childhood but carry a serious prognosis and demand specialist care, and the child who does not respond to a strict diet needs reassessment for these and for inadvertent gluten ingestion, the commonest cause of a non-responsive enteropathy. [12] [11]
Disposition follows the diagnosis and the response. The straightforward case is managed jointly by the general paediatrician or paediatric gastroenterologist and a specialist coeliac dietitian, with the general practitioner supporting annual review and family screening. The child with coeliac crisis, refractory disease, severe malnutrition or a complex associated condition such as type 1 diabetes is managed in a tertiary centre with gastroenterology, dietetics, endocrinology and, where needed, haematology and oncology services. Transition to adult care is planned in mid-adolescence. [11] [4]
Special Populations
The child with type 1 diabetes is the first special population, because the two autoimmune diseases travel together. Routine serological screening at diagnosis and periodically thereafter, using immunoglobulin A anti-tissue-transglutaminase with a total immunoglobulin A, is standard, and a positive screen in an asymptomatic child is treated because of the effects on growth, bone density and glycaemic control. Managing both diseases together demands a dietitian who can reconcile the gluten-free diet with carbohydrate counting and insulin adjustment. [1] [11]
The child with Down syndrome is the second. The relative risk of coeliac disease is several times that of the general population, and the atypical and silent presentations are common, so serological screening is offered even when there are no gut symptoms, repeated periodically through childhood. The same logic applies to Turner syndrome, Williams syndrome, selective immunoglobulin A deficiency and autoimmune thyroiditis, in each of which the background autoimmunity raises the yield of screening and lowers the threshold to test. [4] [6]
The Indigenous, remote and migrant child is the third. In many settings coeliac disease is underdiagnosed in these communities, the diagnostic tests and the gluten-free foods are harder to access, and the burden of untreated malabsorption and faltering growth is carried silently. Culturally safe shared-care pathways, telehealth support for the local team, and attention to the cost and availability of gluten-free alternatives are what make the diagnosis and the diet achievable for families far from a specialist centre. [6] [11]
Evidence, Guidelines & Regional Differences
The evidence base is anchored on the ESPGHAN guideline cycle. The 2012 guideline set the diagnostic framework around serology and biopsy, and the 2020 revision transformed practice by formalising the no-biopsy pathway for the symptomatic child with an immunoglobulin A anti-tissue-transglutaminase titre at or above ten times the upper limit of normal and a positive endomysial antibody on a separate sample, while dropping the earlier human leukocyte antigen and symptom requirements. The 2022 ESPGHAN management position paper then set out the gluten-free diet, the monitoring and the follow-up that close the loop. [1] [2] [11]
The diagnostic accuracy evidence underwrites the serology. The Giersiepen evidence report and the Elwenspoek meta-analysis confirmed the very high sensitivity and specificity of immunoglobulin A anti-tissue-transglutaminase and endomysial antibody in children, and the Werkstetter prospective study validated the no-biopsy approach by showing that a high anti-tissue-transglutaminase titre with a positive endomysial antibody reliably predicts villous atrophy on biopsy. These studies are why the modern pathway can safely spare many children an endoscopy. [5] [9] [8]
The epidemiology and the natural history complete the picture. The Singh global meta-analysis fixed the prevalence at around one per cent and quantified the diagnostic iceberg, while the Lionetti and Crespo-Escobar studies settled the question of gluten timing by showing that neither early nor delayed introduction prevents coeliac disease. The Ludvigsson mortality cohort tied the degree of villous atrophy to a small increase in mortality, which is the long-run argument for a strict lifelong diet, and regional practice differs chiefly in access to the tests and the diet rather than in the diagnostic principle. [6] [7] [12]
Exam Pearls
The no-biopsy pathway — 'TEN-TEN-EMA'
References
- [1]Husby S; Koletzko S; Korponay-Szabó I; Kurppa K; Mearin ML; Ribes-Koninckx C European Society Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J Pediatr Gastroenterol Nutr, 2020.PMID 31568151
- [2]Husby S; Koletzko S; Korponay-Szabó IR; Mearin ML; Phillips A; Shamir R European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr, 2012.PMID 22197856
- [3]Ludvigsson JF; Leffler DA; Bai JC; Biagi F; Fasano A; Green PH The Oslo definitions for coeliac disease and related terms. Gut, 2013.PMID 22345659
- [4]Hill ID; Fasano A; Guandalini S; Hoffenberg E; Levy J; Reilly N NASPGHAN Clinical Report on the Diagnosis and Treatment of Gluten-related Disorders. J Pediatr Gastroenterol Nutr, 2016.PMID 27035374
- [5]Giersiepen K; Lelgemann M; Stuhldreher N; Ronfani L; Husby S; Koletzko S Accuracy of diagnostic antibody tests for coeliac disease in children: summary of an evidence report. J Pediatr Gastroenterol Nutr, 2012.PMID 22266486
- [6]Singh P; Arora A; Strand TA; Leffler DA; Catassi C; Green PH Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol, 2018.PMID 29551598
- [7]Lionetti E; Castellaneta S; Francavilla R; Pulvirenti A; Tonutti E; Amarri S Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med, 2014.PMID 25271602
- [8]Werkstetter KJ; Korponay-Szabó IR; Popp A; Villanacci V; Salemme M; Heilig G Accuracy in Diagnosis of Celiac Disease Without Biopsies in Clinical Practice. Gastroenterology, 2017.PMID 28624578
- [9]Elwenspoek MMC; Jackson J; O'Donnell R; Sinobas A; Dawson S; Everitt H The accuracy of diagnostic indicators for coeliac disease: A systematic review and meta-analysis. PLoS One, 2021.PMID 34695139
- [10]Crespo-Escobar P; Mearin ML; Hervás D; Auricchio R; Castillejo G; Gyimesi J The role of gluten consumption at an early age in celiac disease development: a further analysis of the prospective PreventCD cohort study. Am J Clin Nutr, 2017.PMID 28228423
- [11]Mearin ML; Agardh D; Antunes H; Al-Toma A; Auricchio R; Castillejo G ESPGHAN Position Paper on Management and Follow-up of Children and Adolescents With Celiac Disease. J Pediatr Gastroenterol Nutr, 2022.PMID 35758521
- [12]Ludvigsson JF; Montgomery SM; Ekbom A; Brandt L; Granath F Small-intestinal histopathology and mortality risk in celiac disease. JAMA, 2009.PMID 19755695